lammps vonstress
时间: 2023-07-26 17:02:20 浏览: 146
LAMMPS是一个常用的经典分子动力学模拟软件,通过模拟原子之间的相互作用,可以研究和预测材料的物理和化学性质。其中的vonstress命令是用来计算拉伸应力的工具。以下是对LAMMPS中vonstress命令的解释。
vonstress命令是LAMMPS中的一个计算应力的工具,它通过对原子组进行拉伸,计算并输出应力信息。
在使用vonstress命令之前,需要先定义一个原子组,通常是通过对某一边界进行约束来实现拉伸。例如,可以通过fix命令将原子组约束在某一边界并施加力来实现拉伸。
然后,在拉伸过程中,LAMMPS会自动计算原子组中的应力变化,并使用vonstress命令输出结果。
vonstress命令的输出结果包括应力张量的各个分量和平均应力。应力张量是一个3x3的矩阵,代表了物体在不同方向上的应力情况。vonstress命令输出的应力分量包括xx、yy、zz、xy、xz、yz等,分别代表不同方向上的应力。平均应力则是这些应力分量的平均值。
通过分析vonstress命令输出的结果,我们可以了解到拉伸过程中原子组所处的应力状态。这对于研究材料的力学性质、强度等方面具有重要意义。
总之,vonstress命令是LAMMPS中用于计算拉伸应力的工具,通过对原子组的拉伸,可以获得材料的应力信息。这为材料研究和设计提供了重要的参考依据。
相关问题
lammps molecule
LAMMPS分子动力学模拟软件是一款可用于模拟多种类型物质的分子动力学软件。分子动力学模拟是一种对物质的分子或原子的运动状态进行仿真的方法。LAMMPS模拟软件是一种开源的软件,支持多种平台和操作系统。
通过LAMMPS模拟软件,使用者可以快速构建分子动力学模型,并进行各种分子动力学模拟。模拟器包含数百种已集成好的原子间作用势函数模型,通过这些模型可以快速准确地模拟原子的物理行为。该软件能够模拟各式材料的物理性质,如分子结构、相转换、扩散、断裂、塑性形变等行为。
LAMMPS模拟软件的优点之一是其对各类物质的应用广泛性。它可以模拟单组分、双组分、多组分以及各种类型的材料,如气体、液体、晶体等等。同时,用户可进一步开发新的原子间作用势函数模型,以满足更具体化的模拟要求,从而进一步扩展该软件的模拟领域。
总之,LAMMPS分子动力学模拟软件是一款好用、灵活、可靠的分子动力学软件,其特点是包含大量可定制的功能和模拟算法,以充分满足各种科学计算的需求。它毫无疑问是研究物质微观行为领域的实用工具之一。
lammps colvars
LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator) 是一种用于分子动力学模拟的开源软件包,可用于模拟各种原子和分子系统的动力学行为。COLVARS (Collective Variables) 是 LAMMPS 中的一个插件,用于定义系统的集体变量并对其进行约束,以帮助研究系统的特定性质。
COLVARS 的主要功能是定义和计算系统的集体变量,这些变量是通过原子坐标或其他系统参数来描述的。通过将这些集体变量与力场相结合,COLVARS 可以在模拟过程中实时计算这些变量的值,并将其用于分析和约束。集体变量的定义和计算是灵活可调的,用户可以根据研究的问题选择适当的变量,并使用合适的算法进行计算。
COLVARS 可以用于多种研究方向,包括生物物理、材料科学和化学等。例如,在蛋白质模拟中,可以使用 COLVARS 定义和计算蛋白质的二级结构参数,如氢键数量和二面角。这些集体变量可以用来约束模拟中的结构,以研究蛋白质的稳定性和动力学。
COLVARS 不仅限于集体变量的计算,还可以用于定义约束和谐力场,以约束系统的某些属性,如距离、角度或二面角。这些约束可以用来模拟系统的热力学过程,如相变、溶解和反应等。
总之,LAMMPS 中的 COLVARS 插件提供了一种灵活而强大的工具,用于定义和计算系统的集体变量,并将其用于模拟和分析。通过 COLVARS 的使用,研究人员可以更好地理解和探索原子和分子系统的动力学行为,以及它们与系统性质之间的关联。
阅读全文
相关推荐
大家在看
一种基于SLA的业务管理模型
一种基于SLA的业务管理模型,夏虹,李增智,提出了一种业务级协议(Service Level Agreement,SLA)驱动的业务管理模型,用于将业务使用者和提供者达成的SLA参数转化成设备属性参数。�
Windows_server_2008_R2安装金蝶K3WISE中间层安装与配置。
Windows_server_2008R2安装金蝶K3 WISE中间层安装配置的详细教程讲解。
轻量级xml 解析工具 xml-paras-foxe-CHS.exe
xml_paras_foxe_CHS.exe 轻量级xml 解析工具
信息化综合运维体系.doc
信息化综合运维体系.doc
IMX214_RegisterMap_2.0.0
IMX214_RegisterMap_2.0.0
最新推荐
lammps实例3.pdf
LAMMPS(大型分子多尺度模拟)是一个广泛使用的开源分子动力学软件,适用于模拟从纳米到微米尺度的各种物质,包括软物质和固体物理系统。它的灵活性和高性能使其能够处理当前大多数的势能模型,这得益于其高度优化的...
lammps实例5.pdf
【LAMMPS 简介】 LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)是一款广泛使用的分子动力学模拟软件,它能够处理各种各样的分子和材料模拟问题,包括软物质、生物物理、固体物理等领域。...
lammps实例2.pdf
LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)是一款功能强大的分子模拟软件,它支持多种势能模型,能够高效地处理软材料和固体物理系统的模拟。在本实例中,我们将关注如何使用LAMMPS来...
lammps实例1.pdf
LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)是一款功能强大的分子模拟软件,它支持多种势能模型,如Stillinger-Weber (SW)势,并且具有高效的编程架构,适应于模拟软物质和固体物理系统。...
lammps实例4.pdf
LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)是一款广泛应用于分子模拟的软件,以其高度的编程灵活性和高效的计算性能而闻名。它支持多种势能模型,适用于模拟软物质和固体物理系统。在...
易语言例程:用易核心支持库打造功能丰富的IE浏览框
资源摘要信息:"易语言-易核心支持库实现功能完善的IE浏览框"
易语言是一种简单易学的编程语言,主要面向中文用户。它提供了大量的库和组件,使得开发者能够快速开发各种应用程序。在易语言中,通过调用易核心支持库,可以实现功能完善的IE浏览框。IE浏览框,顾名思义,就是能够在一个应用程序窗口内嵌入一个Internet Explorer浏览器控件,从而实现网页浏览的功能。
易核心支持库是易语言中的一个重要组件,它提供了对IE浏览器核心的调用接口,使得开发者能够在易语言环境下使用IE浏览器的功能。通过这种方式,开发者可以创建一个具有完整功能的IE浏览器实例,它不仅能够显示网页,还能够支持各种浏览器操作,如前进、后退、刷新、停止等,并且还能够响应各种事件,如页面加载完成、链接点击等。
在易语言中实现IE浏览框,通常需要以下几个步骤:
1. 引入易核心支持库:首先需要在易语言的开发环境中引入易核心支持库,这样才能在程序中使用库提供的功能。
2. 创建浏览器控件:使用易核心支持库提供的API,创建一个浏览器控件实例。在这个过程中,可以设置控件的初始大小、位置等属性。
3. 加载网页:将浏览器控件与一个网页地址关联起来,即可在控件中加载显示网页内容。
4. 控制浏览器行为:通过易核心支持库提供的接口,可以控制浏览器的行为,如前进、后退、刷新页面等。同时,也可以响应浏览器事件,实现自定义的交互逻辑。
5. 调试和优化:在开发完成后,需要对IE浏览框进行调试,确保其在不同的操作和网页内容下均能够正常工作。对于性能和兼容性的问题需要进行相应的优化处理。
易语言的易核心支持库使得在易语言环境下实现IE浏览框变得非常方便,它极大地降低了开发难度,并且提高了开发效率。由于易语言的易用性,即使是初学者也能够在短时间内学会如何创建和操作IE浏览框,实现网页浏览的功能。
需要注意的是,由于IE浏览器已经逐渐被微软边缘浏览器(Microsoft Edge)所替代,使用IE核心的技术未来可能面临兼容性和安全性的挑战。因此,在实际开发中,开发者应考虑到这一点,并根据需求选择合适的浏览器控件实现技术。
此外,易语言虽然简化了编程过程,但其在功能上可能不如主流的编程语言(如C++, Java等)强大,且社区和技术支持相比其他语言可能较为有限,这些都是在选择易语言作为开发工具时需要考虑的因素。
文件名列表中的“IE类”可能是指包含实现IE浏览框功能的类库或者示例代码。在易语言中,类库是一组封装好的代码模块,其中包含了各种功能的实现。通过在易语言项目中引用这些类库,开发者可以简化开发过程,快速实现特定功能。而示例代码则为开发者提供了具体的实现参考,帮助理解和学习如何使用易核心支持库来创建IE浏览框。
管理建模和仿真的文件
管理Boualem Benatallah引用此版本:布阿利姆·贝纳塔拉。管理建模和仿真。约瑟夫-傅立叶大学-格勒诺布尔第一大学,1996年。法语。NNT:电话:00345357HAL ID:电话:00345357https://theses.hal.science/tel-003453572008年12月9日提交HAL是一个多学科的开放存取档案馆,用于存放和传播科学研究论文,无论它们是否被公开。论文可以来自法国或国外的教学和研究机构,也可以来自公共或私人研究中心。L’archive ouverte pluridisciplinaire
STM32F407ZG引脚功能深度剖析:掌握引脚分布与配置的秘密(全面解读)
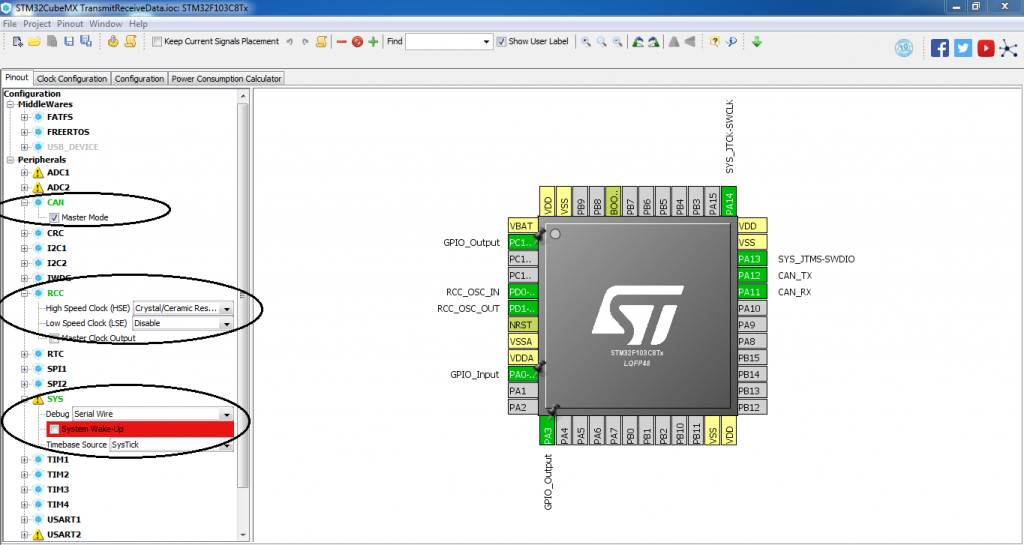
# 摘要
本文全面介绍了STM32F407ZG微控制器的引脚特性、功能、配置和应用。首先概述了该芯片的引脚布局,然后详细探讨了标准外设、高级控制以及特殊功能引脚的不同配置和使用方法。在此基础上,文章深入分析了引脚模式配置、高级配置技巧,并提供了实际应用案例,如LED控制和串口通信。在设计方面,阐述了引脚布局策略、多层板设计及高密度引脚应用的解决方案。最后,介绍
给出文档中问题的答案代码
您提到的是需要编写MATLAB代码来实现文档中的实验任务。以下是根据文档内容编写的MATLAB代码示例:
```matlab
% 上机2 实验代码
% 读取输入图像
inputImage = imread('your_face_image.jpg'); % 替换为您的图像文件路径
if size(inputImage, 1) < 1024 || size(inputImage, 2) < 1024
error('图像尺寸必须大于1024x1024');
end
% 将彩色图像转换为灰度图像
grayImage = rgb2gray(inputImage);
% 调整图像大小为5
Docker构建与运行Next.js应用的指南
资源摘要信息:"rivoltafilippo-next-main"
在探讨“rivoltafilippo-next-main”这一资源时,首先要从标题“rivoltafilippo-next”入手。这个标题可能是某一项目、代码库或应用的命名,结合描述中提到的Docker构建和运行命令,我们可以推断这是一个基于Docker的Node.js应用,特别是使用了Next.js框架的项目。Next.js是一个流行的React框架,用于服务器端渲染和静态网站生成。
描述部分提供了构建和运行基于Docker的Next.js应用的具体命令:
1. `docker build`命令用于创建一个新的Docker镜像。在构建镜像的过程中,开发者可以定义Dockerfile文件,该文件是一个文本文件,包含了创建Docker镜像所需的指令集。通过使用`-t`参数,用户可以为生成的镜像指定一个标签,这里的标签是`my-next-js-app`,意味着构建的镜像将被标记为`my-next-js-app`,方便后续的识别和引用。
2. `docker run`命令则用于运行一个Docker容器,即基于镜像启动一个实例。在这个命令中,`-p 3000:3000`参数指示Docker将容器内的3000端口映射到宿主机的3000端口,这样做通常是为了让宿主机能够访问容器内运行的应用。`my-next-js-app`是容器运行时使用的镜像名称,这个名称应该与构建时指定的标签一致。
最后,我们注意到资源包含了“TypeScript”这一标签,这表明项目可能使用了TypeScript语言。TypeScript是JavaScript的一个超集,它添加了静态类型定义的特性,能够帮助开发者更容易地维护和扩展代码,尤其是在大型项目中。
结合资源名称“rivoltafilippo-next-main”,我们可以推测这是项目的主目录或主仓库。通常情况下,开发者会将项目的源代码、配置文件、构建脚本等放在一个主要的目录中,这个目录通常命名为“main”或“src”等,以便于管理和维护。
综上所述,我们可以总结出以下几个重要的知识点:
- Docker容器和镜像的概念以及它们之间的关系:Docker镜像是静态的只读模板,而Docker容器是从镜像实例化的动态运行环境。
- `docker build`命令的使用方法和作用:这个命令用于创建新的Docker镜像,通常需要一个Dockerfile来指定构建的指令和环境。
- `docker run`命令的使用方法和作用:该命令用于根据镜像启动一个或多个容器实例,并可指定端口映射等运行参数。
- Next.js框架的特点:Next.js是一个支持服务器端渲染和静态网站生成的React框架,适合构建现代的Web应用。
- TypeScript的作用和优势:TypeScript是JavaScript的一个超集,它提供了静态类型检查等特性,有助于提高代码质量和可维护性。
- 项目资源命名习惯:通常项目会有一个主目录,用来存放项目的源代码和核心配置文件,以便于项目的版本控制和团队协作。
以上内容基于给定的信息进行了深入的分析,为理解该项目的构建、运行方式以及技术栈提供了基础。在实际开发中,开发者应当参考更详细的文档和指南,以更高效地管理和部署基于Docker和TypeScript的Next.js项目。