tblastn2.12.0+
时间: 2023-10-01 19:00:41 浏览: 40
tblastn2.12.0是一种用于进行蛋白质与DNA序列的比对和相似性分析的工具。它是NCBI(美国国家生物技术信息中心)提供的一种基于BLAST(Basic Local Alignment Search Tool,基本局部序列比对工具)算法的软件。tblastn2.12.0的主要功能是在数据库中搜索DNA序列与已知蛋白质序列之间的相似性。
tblastn2.12.0在进行蛋白质与DNA序列比对时,采用的是蛋白质序列作为查询序列,而DNA序列作为参考数据库。通过比对查询序列与数据库中的DNA序列,可以找到两者之间的相似性,判断某个DNA序列是否可能编码某个蛋白质。
tblastn2.12.0具有高效、准确的特点,可以在大规模基因组数据库中快速查找到与查询序列相似的DNA序列。它不仅可以发现已知蛋白质的同源序列,还能够发现新的蛋白质序列和结构域。
tblastn2.12.0的使用非常广泛,是生物信息学领域中进行蛋白质与DNA序列比对和相似性分析的重要工具。研究人员可以利用tblastn2.12.0来研究蛋白质的进化、功能和结构等特性,从而深入理解生物体的分子机制。
相关问题
blast怎么用 要命令
BLAST (Basic Local Alignment Search Tool) 是一种在生物信息学研究中广泛使用的工具,用于在生物序列数据库中搜索相似序列。常用的BLAST命令包括:
1. 搜索核酸序列数据库:
```
blastn -query input.fasta -db database_name -out output.txt
```
2. 搜索蛋白质序列数据库:
```
blastp -query input.fasta -db database_name -out output.txt
```
3. 搜索核酸序列在蛋白质数据库中的同源序列:
```
blastx -query input.fasta -db database_name -out output.txt
```
4. 搜索蛋白质序列在核酸数据库中的同源序列:
```
tblastn -query input.fasta -db database_name -out output.txt
```
以上命令中,`input.fasta` 是输入序列文件,`database_name` 是要搜索的数据库名称,`output.txt` 是输出结果文件。还可以使用其他参数来控制BLAST的搜索行为,如设置阈值和过滤器等。
blast比对1500个基因用什么参数
对于BLAST比对,参数的选择应该根据具体的情况进行调整,包括待比对的序列长度、比对的数据库、比对的算法、期望的匹配程度等等。对于比对1500个基因的情况,一些常用的参数如下:
1. 比对算法选择:对于长序列或者比对的序列相似度较低的情况,可以选择BLASTN算法进行比对。对于比对的蛋白质序列,可以选择BLASTP算法进行比对。
2. 数据库选择:可以选择NCBI的NT数据库或者NR数据库进行比对。也可以根据具体的需求选择其他数据库。
3. 期望的匹配程度(E value):E value越小,表示匹配的可信度越高。可以根据具体的需求调整E value的值。
4. 比对的阈值(cutoff):可以设置一个比对的阈值,只有得分高于该阈值的比对结果才会被保留。
5. 多序列比对(MSA):如果需要进行多个序列的比对,可以选择BLASTX或者TBLASTN算法进行比对。
需要注意的是,参数的选择应该根据具体的情况进行调整,不同的参数设置会对比对结果产生不同的影响。
相关推荐
最新推荐
华为OD机试D卷 - 用连续自然数之和来表达整数 - 免费看解析和代码.html
私信博主免费获取真题解析以及代码
Screenshot_2024-05-10-20-21-01-857_com.chaoxing.mobile.jpg
Screenshot_2024-05-10-20-21-01-857_com.chaoxing.mobile.jpg
数字图像处理|Matlab-频域增强实验-彩色图像的频域滤波.zip
数字图像处理|Matlab-频域增强实验-彩色图像的频域滤波.zip
2024-2030中国定向转向膜市场现状研究分析与发展前景预测报告.docx
2024-2030中国定向转向膜市场现状研究分析与发展前景预测报告
开源工时填报管理系统安装包
开源工时填报管理系统安装包
zigbee-cluster-library-specification
最新的zigbee-cluster-library-specification说明文档。
管理建模和仿真的文件
管理Boualem Benatallah引用此版本:布阿利姆·贝纳塔拉。管理建模和仿真。约瑟夫-傅立叶大学-格勒诺布尔第一大学,1996年。法语。NNT:电话:00345357HAL ID:电话:00345357https://theses.hal.science/tel-003453572008年12月9日提交HAL是一个多学科的开放存取档案馆,用于存放和传播科学研究论文,无论它们是否被公开。论文可以来自法国或国外的教学和研究机构,也可以来自公共或私人研究中心。L’archive ouverte pluridisciplinaire
实现实时数据湖架构:Kafka与Hive集成
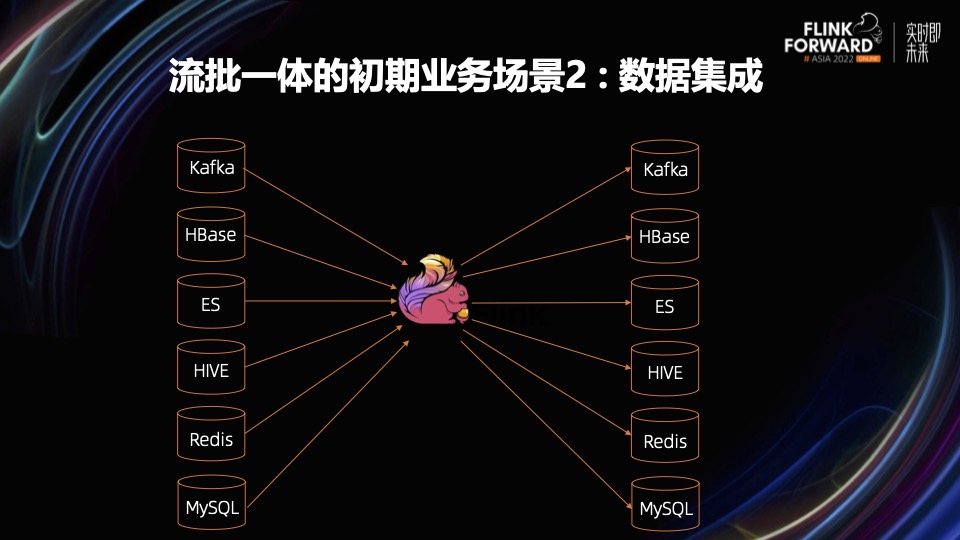
# 1. 实时数据湖架构概述**
实时数据湖是一种现代数据管理架构,它允许企业以低延迟的方式收集、存储和处理大量数据。与传统数据仓库不同,实时数据湖不依赖于预先定义的模式,而是采用灵活的架构,可以处理各种数据类型和格式。这种架构为企业提供了以下优势:
- **实时洞察:**实时数据湖允许企业访问最新的数据,从而做出更明智的决策。
- **数据民主化:**实时数据湖使各种利益相关者都可
spring添加xml配置文件
1. 创建一个新的Spring配置文件,例如"applicationContext.xml"。
2. 在文件头部添加XML命名空间和schema定义,如下所示:
```
<beans xmlns="http://www.springframework.org/schema/beans"
xmlns:xsi="http://www.w3.org/2001/XMLSchema-instance"
xsi:schemaLocation="http://www.springframework.org/schema/beans
JSBSim Reference Manual
JSBSim参考手册,其中包含JSBSim简介,JSBSim配置文件xml的编写语法,编程手册以及一些应用实例等。其中有部分内容还没有写完,估计有生之年很难看到完整版了,但是内容还是很有参考价值的。