python基因富集
时间: 2023-10-15 22:28:07 浏览: 59
基因富集是指在一组基因中寻找具有特定功能或参与特定生物学过程的基因的统计显著性增加。在Python中,有一些常用的工具可以用于基因富集分析。以下是一些常见的Python库和工具:
1. Enrichr:一个在线基因富集分析工具,提供了Python API,可以通过Enrichr API访问并获取富集结果。
2. gseapy:一个基因富集分析的Python库,可以用于富集分析、可视化和结果解释等。
3. clusterProfiler:一个R包,但也有Python的接口。它提供了丰富的功能用于功能注释和富集分析。
4. GOATOOLS:一个用于基因本体(Gene Ontology)富集分析的Python库,可以用于基因集的GO注释和富集分析。
这些工具可以帮助您进行基因富集分析,并提供了相应的函数和方法来计算基因组中的显著富集。您可以根据您的需求选择适合您的工具,并根据其文档和示例来使用它们。
相关问题
python的生物信息
Python是生物信息学领域中最流行的编程语言之一,它具有易学易用、灵活性强、生态系统丰富等特点,因此被广泛应用于生物信息学的数据处理、分析、建模和可视化等方面。
以下是Python在生物信息学中的常见应用:
1.生物序列分析。使用Python的生物信息学库,例如Biopython,可以对生物序列进行处理、分析、比对、注释、可视化等操作。例如,可以使用Biopython提供的Seq对象来操作DNA、RNA和蛋白质序列,使用SeqRecord对象来处理序列注释信息,使用BLAST、Clustal等工具来进行序列比对和多序列比对等。
2.基因表达分析。使用Python可以处理RNA-Seq、ChIP-Seq、ATAC-Seq等高通量测序数据,例如进行差异表达分析、富集分析、基因网络分析等。常用的生物信息学库包括DESeq2、edgeR、limma等。
3.蛋白质结构预测和分析。使用Python可以进行蛋白质的结构预测、蛋白质复合物的分析、蛋白质动力学模拟等操作。常用的生物信息学库包括PyMOL、MDAnalysis、ProDy等。
4.系统生物学建模。使用Python可以进行基于系统生物学的建模和仿真,并对生物系统的动态行为进行分析。常用的生物信息学库包括PySB、COPASI、Tellurium等。
总之,Python在生物信息学中的应用非常广泛,如果您想学习Python在生物信息学中的应用,可以先学习Python的基础语法,然后深入学习一些生物信息学库和工具,例如Biopython、DESeq2等。另外,参加一些生物信息学的在线课程和培训也是一个不错的选择。
ssgsea富集分析代码
SSGSEA(Single-sample Gene Set Enrichment Analysis)是一种基于基因集富集分析的方法,可以对单个样本进行基因表达谱的分析。以下是一个Python实现的SSGSEA富集分析代码示例:
```python
import numpy as np
from scipy.stats import norm
def ssgsea(gene_exp, gene_sets, nperm=1000, weighted_score_type=1, permutation=True,
min_size=1, max_size=5000, verbose=False, seed=None):
"""
:param gene_exp: array-like, shape (n_samples, n_features)
Gene expression matrix (rows are samples and columns are features).
:param gene_sets: dict
Gene sets in the format of dictionary. Keys are pathway names and values are gene lists.
:param nperm: int, optional
The number of permutations for calculating the p-value. Default is 1000.
:param weighted_score_type: int, optional
The weighting score type. Default is 1.
:param permutation: bool, optional
Whether to do permutation for calculating the p-value. Default is True.
:param min_size: int, optional
The minimum number of genes in a gene set to be considered. Default is 1.
:param max_size: int, optional
The maximum number of genes in a gene set to be considered. Default is 5000.
:param verbose: bool, optional
Whether to print the progress. Default is False.
:param seed: int, optional
The seed for the random number generator. Default is None.
:return: dict
A dictionary of pathway names and enrichment scores.
"""
# Initialize the random number generator
if seed is not None:
np.random.seed(seed)
# Prepare the gene expression matrix
gene_exp = np.array(gene_exp)
# Prepare the gene set list
gene_sets = {k: v for k, v in gene_sets.items() if min_size <= len(v) <= max_size}
# Compute the gene set scores
pathways = {}
for pathway, genes in gene_sets.items():
# Compute the gene set score for each sample
gss = []
for i in range(gene_exp.shape[0]):
# Get the gene expression values for the pathway genes
pathway_exp = gene_exp[i, np.isin(gene_exp.columns, genes)]
# Compute the gene set score
if weighted_score_type == 0:
gss.append(pathway_exp.sum())
elif weighted_score_type == 1:
gss.append(pathway_exp.mean())
elif weighted_score_type == -1:
gss.append(pathway_exp.abs().mean())
# Compute the enrichment score and p-value
if permutation:
null_gss = []
for i in range(nperm):
# Shuffle the gene expression values
shuffle_exp = gene_exp.apply(np.random.permutation, axis=1)
# Compute the gene set score for each sample
null_gss.append(shuffle_exp.apply(lambda x: x[np.isin(gene_exp.columns, genes)].mean(), axis=1))
null_gss = pd.concat(null_gss, axis=1)
null_es = null_gss.apply(lambda x: (x > gss).mean() - (x < gss).mean())
es = (gss - null_es.mean()) / null_es.std()
pval = (null_es < gss).mean()
else:
es = (gss - gss.mean()) / gss.std()
pval = 1 - norm.cdf(es)
pathways[pathway] = {'es': es, 'pval': pval}
if verbose:
print('%s: ES = %.3f, p-value = %.3f' % (pathway, es, pval))
return pathways
```
该代码使用了NumPy和SciPy库进行计算。在使用时,需要将基因表达谱和基因集传递给`ssgsea`函数。此外,还可以设置其他参数,例如是否进行置换和置换次数等。函数返回一个包含富集分析结果的字典。
相关推荐
最新推荐
Python tkinter模版代码实例
主要介绍了Python tkinter模版代码实例,文中通过示例代码介绍的非常详细,对大家的学习或者工作具有一定的参考学习价值,需要的朋友可以参考下
python url 参数修改方法
今天小编就为大家分享一篇python url 参数修改方法,具有很好的参考价值,希望对大家有所帮助。一起跟随小编过来看看吧
Python使用OpenCV进行标定
主要介绍了Python使用OpenCV进行标定,小编觉得挺不错的,现在分享给大家,也给大家做个参考。一起跟随小编过来看看吧
QPSK调制原理及python实现
文章目录QPSK调制原理及python实现QPSK调制原理python实现调制过程1、导入相关库函数2、调制过程3、作图过程 QPSK调制原理及python实现 QPSK调制原理 QPSK调制过程及原理在前面的博客中以及详细分析过。在本文中将...
python距离测量的方法
主要为大家详细介绍了python距离测量的方法,具有一定的参考价值,感兴趣的小伙伴们可以参考一下
zigbee-cluster-library-specification
最新的zigbee-cluster-library-specification说明文档。
管理建模和仿真的文件
管理Boualem Benatallah引用此版本:布阿利姆·贝纳塔拉。管理建模和仿真。约瑟夫-傅立叶大学-格勒诺布尔第一大学,1996年。法语。NNT:电话:00345357HAL ID:电话:00345357https://theses.hal.science/tel-003453572008年12月9日提交HAL是一个多学科的开放存取档案馆,用于存放和传播科学研究论文,无论它们是否被公开。论文可以来自法国或国外的教学和研究机构,也可以来自公共或私人研究中心。L’archive ouverte pluridisciplinaire
实现实时数据湖架构:Kafka与Hive集成
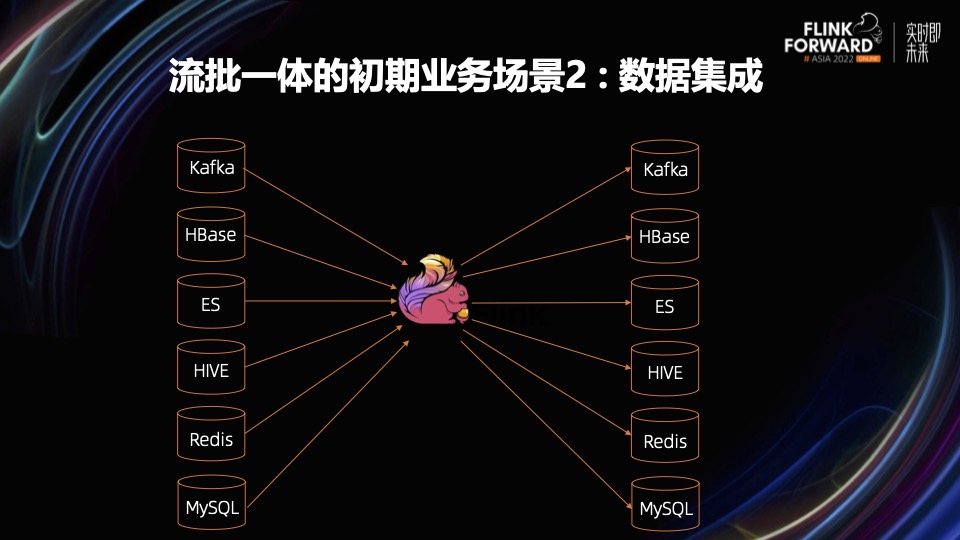
# 1. 实时数据湖架构概述**
实时数据湖是一种现代数据管理架构,它允许企业以低延迟的方式收集、存储和处理大量数据。与传统数据仓库不同,实时数据湖不依赖于预先定义的模式,而是采用灵活的架构,可以处理各种数据类型和格式。这种架构为企业提供了以下优势:
- **实时洞察:**实时数据湖允许企业访问最新的数据,从而做出更明智的决策。
- **数据民主化:**实时数据湖使各种利益相关者都可
如何用python编写api接口
在Python中编写API接口可以使用多种框架,其中比较流行的有Flask和Django。这里以Flask框架为例,简单介绍如何编写API接口。
1. 安装Flask框架
使用pip命令安装Flask框架:
```
pip install flask
```
2. 编写API接口
创建一个Python文件,例如app.py,编写以下代码:
```python
from flask import Flask, jsonify
app = Flask(__name__)
@app.route('/api/hello', methods=['GET'])
def hello():
JSBSim Reference Manual
JSBSim参考手册,其中包含JSBSim简介,JSBSim配置文件xml的编写语法,编程手册以及一些应用实例等。其中有部分内容还没有写完,估计有生之年很难看到完整版了,但是内容还是很有参考价值的。