使用r创建基于基因网络的可视化
时间: 2023-11-20 10:02:38 浏览: 35
在R语言中,我们可以使用不同的包和工具来创建基于基因网络的可视化。
首先,我们可以使用Bioconductor包中的“igraph”来构建和可视化基因网络。该包提供了一个用于分析和操作图形数据的强大框架。我们可以使用该包中的函数来创建一个空白的网络图,并使用基因之间的相互作用来添加边缘。通过设置边的属性,我们可以将基因之间的关系表示为权重。然后,我们可以使用绘图函数将网络可视化为一个图形。
其次,我们还可以使用“networkD3”包来创建基于基因网络的可视化。这个包提供了一些用于创建交互式网络图的函数。我们可以使用自己的基因网络数据来创建一个网络对象,并使用不同的布局算法将基因表示为节点,并使用他们之间的相互作用来添加边。然后,我们可以使用绘图函数将网络呈现为一个交互式图形,用户可以点击和拖动来探索网络中的关系。
此外,我们还可以使用“ggplot2”包来创建基于基因网络的可视化。虽然ggplot2主要用于绘制统计图形,但我们可以使用它的强大功能来创建一个自定义的网络图。我们可以使用ggplot2中的几何函数来绘制节点和边,并使用aes函数将基因之间的关系映射到图形的属性,如节点的颜色或边的宽度。然后,我们可以使用主题和标签函数来增强图形的可读性和美观性。
总之,使用R语言,我们可以使用不同的包和工具来创建基于基因网络的可视化。这些可视化有助于理解基因之间的相互作用和关系,并为基因组学研究提供有价值的洞察。
相关问题
如何使用R将细菌基因序列可视化
您可以使用R中的ggplot2包和Biostrings包来可视化细菌基因序列。以下是一个简单的步骤:
1.首先,您需要从NCBI数据库中下载细菌基因组序列,并将其导入到R中。
2.使用Biostrings包中的readDNAStringSet函数将基因序列读入到R中。例如:
```
library(Biostrings)
genome <- readDNAStringSet("bacteria_genome.fasta")
```
3.使用ggplot2包中的geom_segment函数和基因序列的位置信息来绘制基因组图。例如:
```
library(ggplot2)
ggplot() +
geom_segment(aes(x=start, y=1, xend=end, yend=1), data=as.data.frame(genome), color="black", size=0.5) +
scale_x_continuous(limits=c(0, length(genome))) +
theme_void()
```
这将绘制一条黑色的线段,表示整个基因组序列。
4.使用不同的颜色和形状绘制不同的基因和基因组元素。例如:
```
genes <- data.frame(name=c("gene1", "gene2"), start=c(100, 500), end=c(200, 600))
ggplot() +
geom_segment(aes(x=start, y=1, xend=end, yend=1), data=as.data.frame(genome), color="black", size=0.5) +
geom_point(aes(x=start, y=1, shape=name), data=genes, color="blue", size=3) +
geom_point(aes(x=end, y=1, shape=name), data=genes, color="red", size=3) +
scale_x_continuous(limits=c(0, length(genome))) +
theme_void()
```
这将绘制两个蓝色和红色的点,分别表示两个基因的起始和终止位置,并使用不同的形状表示它们的名称。
您可以根据需要进行更多的调整和修改,以获得更好的可视化效果。
使用R将细菌基因序列可视化的具体步骤
下面是使用R将细菌基因序列可视化的具体步骤:
1. 导入基因序列数据
首先需要将基因序列数据导入到R中。可以使用read.fasta()函数来读取fasta格式的序列文件。假设我们的基因序列文件名为“gene.fasta”,那么可以使用以下代码将其导入到R中:
```
library(seqinr)
gene_seq <- read.fasta("gene.fasta")
```
2. 特征提取
接下来需要对基因序列进行特征提取,常见的特征包括GC含量、碱基长度、开放阅读框等。假设我们要提取GC含量和碱基长度这两个特征,可以使用以下代码:
```
# GC含量
gc_content <- sapply(gene_seq, function(x) {
gc <- sum(x == "G" | x == "C")
at <- sum(x == "A" | x == "T")
gc / (gc + at)
})
# 碱基长度
base_length <- sapply(gene_seq, length)
```
在上面的代码中,我们使用sapply()函数将函数应用到每条基因序列上,然后计算出GC含量和碱基长度。
3. 可视化处理
接下来就可以使用ggplot2包中的函数进行可视化处理了。以下是绘制GC含量图表的代码:
```
library(ggplot2)
df_gc <- data.frame(gc = gc_content)
ggplot(df_gc, aes(x = gc)) +
geom_density(fill = "blue") +
labs(x = "GC Content", y = "Density", title = "GC Content Distribution")
```
在上面的代码中,我们将GC含量数据转换为数据框,并使用ggplot()函数创建一个图形对象。然后使用geom_density()函数绘制密度曲线,并使用labs()函数添加标题和轴标签。
以下是绘制碱基长度变化图表的代码:
```
df_length <- data.frame(length = base_length)
ggplot(df_length, aes(x = length)) +
geom_histogram(binwidth = 500, fill = "red") +
labs(x = "Base Length", y = "Frequency", title = "Base Length Distribution")
```
在上面的代码中,我们将碱基长度数据转换为数据框,并使用ggplot()函数创建一个图形对象。然后使用geom_histogram()函数绘制直方图,并使用labs()函数添加标题和轴标签。
4. 图表美化
最后,我们可以对图表进行美化,例如添加标题、更改颜色、调整字体大小等。以下是对GC含量图表进行美化的代码:
```
ggplot(df_gc, aes(x = gc)) +
geom_density(fill = "blue") +
labs(x = "GC Content", y = "Density", title = "GC Content Distribution") +
theme(plot.title = element_text(size = 18, face = "bold"),
axis.title = element_text(size = 14, face = "bold"),
axis.text = element_text(size = 12))
```
在上面的代码中,我们使用theme()函数对图表进行美化,例如增加标题字体大小、轴标签字体大小和轴刻度字体大小等。
以上就是使用R将细菌基因序列可视化的具体步骤。
相关推荐
最新推荐
基于vue+echarts 数据可视化大屏展示的方法示例
主要介绍了基于vue+echarts 数据可视化大屏展示的方法示例,小编觉得挺不错的,现在分享给大家,也给大家做个参考。一起跟随小编过来看看吧
使用pytorch实现可视化中间层的结果
今天小编就为大家分享一篇使用pytorch实现可视化中间层的结果,具有很好的参考价值,希望对大家有所帮助。一起跟随小编过来看看吧
python使用pyecharts库画地图数据可视化的实现
主要介绍了python使用pyecharts库画地图数据可视化的实现,文中通过示例代码介绍的非常详细,对大家的学习或者工作具有一定的参考学习价值,需要的朋友们下面随着小编来一起学习学习吧
Echarts可视化工具的使用案例(idea)
Echart可视化工具的简单实用,本文章采用idea开发环境进行案例实施,运用到javaEE、ajax、serverlet技术
Django上使用数据可视化利器Bokeh解析
主要介绍了Django上使用数据可视化利器Bokeh解析,文中通过示例代码介绍的非常详细,对大家的学习或者工作具有一定的参考学习价值,需要的朋友可以参考下
zigbee-cluster-library-specification
最新的zigbee-cluster-library-specification说明文档。
管理建模和仿真的文件
管理Boualem Benatallah引用此版本:布阿利姆·贝纳塔拉。管理建模和仿真。约瑟夫-傅立叶大学-格勒诺布尔第一大学,1996年。法语。NNT:电话:00345357HAL ID:电话:00345357https://theses.hal.science/tel-003453572008年12月9日提交HAL是一个多学科的开放存取档案馆,用于存放和传播科学研究论文,无论它们是否被公开。论文可以来自法国或国外的教学和研究机构,也可以来自公共或私人研究中心。L’archive ouverte pluridisciplinaire
实现实时数据湖架构:Kafka与Hive集成
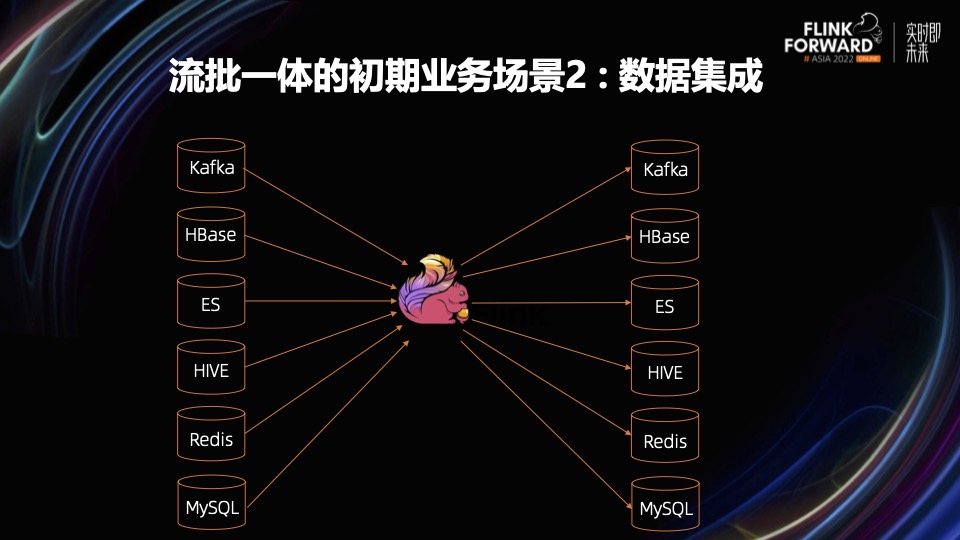
# 1. 实时数据湖架构概述**
实时数据湖是一种现代数据管理架构,它允许企业以低延迟的方式收集、存储和处理大量数据。与传统数据仓库不同,实时数据湖不依赖于预先定义的模式,而是采用灵活的架构,可以处理各种数据类型和格式。这种架构为企业提供了以下优势:
- **实时洞察:**实时数据湖允许企业访问最新的数据,从而做出更明智的决策。
- **数据民主化:**实时数据湖使各种利益相关者都可
spring添加xml配置文件
1. 创建一个新的Spring配置文件,例如"applicationContext.xml"。
2. 在文件头部添加XML命名空间和schema定义,如下所示:
```
<beans xmlns="http://www.springframework.org/schema/beans"
xmlns:xsi="http://www.w3.org/2001/XMLSchema-instance"
xsi:schemaLocation="http://www.springframework.org/schema/beans
JSBSim Reference Manual
JSBSim参考手册,其中包含JSBSim简介,JSBSim配置文件xml的编写语法,编程手册以及一些应用实例等。其中有部分内容还没有写完,估计有生之年很难看到完整版了,但是内容还是很有参考价值的。