使用R将细菌基因序列可视化的具体步骤
时间: 2023-12-29 19:05:56 浏览: 28
下面是使用R将细菌基因序列可视化的具体步骤:
1. 导入基因序列数据
首先需要将基因序列数据导入到R中。可以使用read.fasta()函数来读取fasta格式的序列文件。假设我们的基因序列文件名为“gene.fasta”,那么可以使用以下代码将其导入到R中:
```
library(seqinr)
gene_seq <- read.fasta("gene.fasta")
```
2. 特征提取
接下来需要对基因序列进行特征提取,常见的特征包括GC含量、碱基长度、开放阅读框等。假设我们要提取GC含量和碱基长度这两个特征,可以使用以下代码:
```
# GC含量
gc_content <- sapply(gene_seq, function(x) {
gc <- sum(x == "G" | x == "C")
at <- sum(x == "A" | x == "T")
gc / (gc + at)
})
# 碱基长度
base_length <- sapply(gene_seq, length)
```
在上面的代码中,我们使用sapply()函数将函数应用到每条基因序列上,然后计算出GC含量和碱基长度。
3. 可视化处理
接下来就可以使用ggplot2包中的函数进行可视化处理了。以下是绘制GC含量图表的代码:
```
library(ggplot2)
df_gc <- data.frame(gc = gc_content)
ggplot(df_gc, aes(x = gc)) +
geom_density(fill = "blue") +
labs(x = "GC Content", y = "Density", title = "GC Content Distribution")
```
在上面的代码中,我们将GC含量数据转换为数据框,并使用ggplot()函数创建一个图形对象。然后使用geom_density()函数绘制密度曲线,并使用labs()函数添加标题和轴标签。
以下是绘制碱基长度变化图表的代码:
```
df_length <- data.frame(length = base_length)
ggplot(df_length, aes(x = length)) +
geom_histogram(binwidth = 500, fill = "red") +
labs(x = "Base Length", y = "Frequency", title = "Base Length Distribution")
```
在上面的代码中,我们将碱基长度数据转换为数据框,并使用ggplot()函数创建一个图形对象。然后使用geom_histogram()函数绘制直方图,并使用labs()函数添加标题和轴标签。
4. 图表美化
最后,我们可以对图表进行美化,例如添加标题、更改颜色、调整字体大小等。以下是对GC含量图表进行美化的代码:
```
ggplot(df_gc, aes(x = gc)) +
geom_density(fill = "blue") +
labs(x = "GC Content", y = "Density", title = "GC Content Distribution") +
theme(plot.title = element_text(size = 18, face = "bold"),
axis.title = element_text(size = 14, face = "bold"),
axis.text = element_text(size = 12))
```
在上面的代码中,我们使用theme()函数对图表进行美化,例如增加标题字体大小、轴标签字体大小和轴刻度字体大小等。
以上就是使用R将细菌基因序列可视化的具体步骤。
相关推荐
最新推荐
微信小程序-番茄时钟源码
微信小程序番茄时钟的源码,支持进一步的修改。番茄钟,指的是把工作任务分解成半小时左右,集中精力工作25分钟后休息5分钟,如此视作种一个“番茄”,而“番茄工作法”的流程能使下一个30分钟更有动力。
激光雷达专题研究:迈向高阶智能化关键,前瞻布局把握行业脉搏.pdf
电子元件 电子行业 行业分析 数据分析 数据报告 行业报告
安享智慧理财测试项目Mock服务代码
安享智慧理财测试项目Mock服务代码
课程设计 基于SparkMLlib的ALS算法的电影推荐系统源码+详细文档+全部数据齐全.zip
【资源说明】
课程设计 基于SparkMLlib的ALS算法的电影推荐系统源码+详细文档+全部数据齐全.zip课程设计 基于SparkMLlib的ALS算法的电影推荐系统源码+详细文档+全部数据齐全.zip
【备注】
1、该项目是高分毕业设计项目源码,已获导师指导认可通过,答辩评审分达到95分
2、该资源内项目代码都经过测试运行成功,功能ok的情况下才上传的,请放心下载使用!
3、本项目适合计算机相关专业(如软件工程、计科、人工智能、通信工程、自动化、电子信息等)的在校学生、老师或者企业员工下载使用,也可作为毕业设计、课程设计、作业、项目初期立项演示等,当然也适合小白学习进阶。
4、如果基础还行,可以在此代码基础上进行修改,以实现其他功能,也可直接用于毕设、课设、作业等。
欢迎下载,沟通交流,互相学习,共同进步!
华中科技大学电信专业 课程资料 作业 代码 实验报告-雷达与信息对抗-内含源码和说明书.zip
华中科技大学电信专业 课程资料 作业 代码 实验报告-雷达与信息对抗-内含源码和说明书.zip
zigbee-cluster-library-specification
最新的zigbee-cluster-library-specification说明文档。
管理建模和仿真的文件
管理Boualem Benatallah引用此版本:布阿利姆·贝纳塔拉。管理建模和仿真。约瑟夫-傅立叶大学-格勒诺布尔第一大学,1996年。法语。NNT:电话:00345357HAL ID:电话:00345357https://theses.hal.science/tel-003453572008年12月9日提交HAL是一个多学科的开放存取档案馆,用于存放和传播科学研究论文,无论它们是否被公开。论文可以来自法国或国外的教学和研究机构,也可以来自公共或私人研究中心。L’archive ouverte pluridisciplinaire
实现实时数据湖架构:Kafka与Hive集成
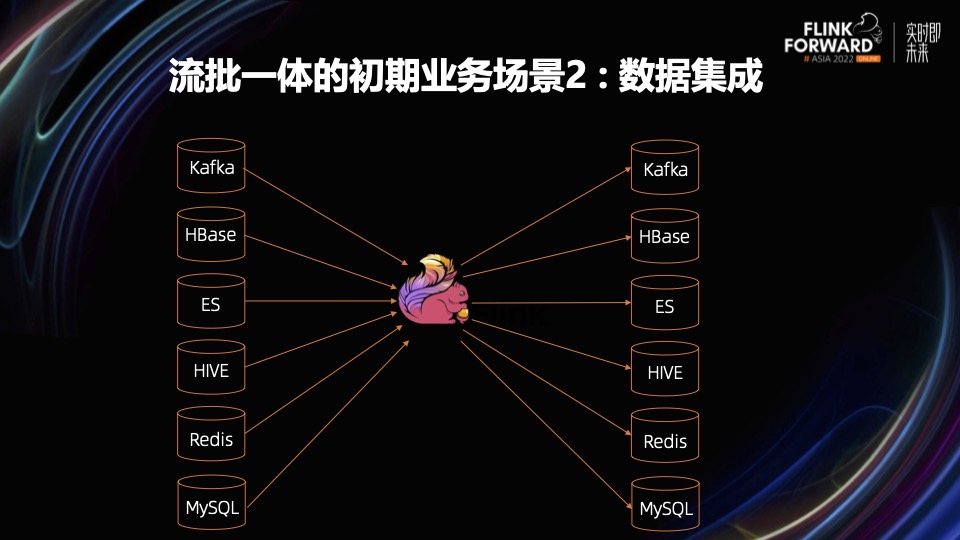
# 1. 实时数据湖架构概述**
实时数据湖是一种现代数据管理架构,它允许企业以低延迟的方式收集、存储和处理大量数据。与传统数据仓库不同,实时数据湖不依赖于预先定义的模式,而是采用灵活的架构,可以处理各种数据类型和格式。这种架构为企业提供了以下优势:
- **实时洞察:**实时数据湖允许企业访问最新的数据,从而做出更明智的决策。
- **数据民主化:**实时数据湖使各种利益相关者都可
用 Python 画一个可以动的爱心
好的,我可以帮助您使用Python来绘制一个动态的爱心。您可以使用turtle库来实现。以下是实现代码:
```python
import turtle
import math
# 设置画布和画笔
canvas = turtle.Screen()
canvas.bgcolor("black")
pencil = turtle.Turtle()
pencil.speed(0)
pencil.color("red", "pink")
pencil.pensize(3)
# 定义爱心函数
def draw_love(heart_size, x_offset=0, y_offset=0):
JSBSim Reference Manual
JSBSim参考手册,其中包含JSBSim简介,JSBSim配置文件xml的编写语法,编程手册以及一些应用实例等。其中有部分内容还没有写完,估计有生之年很难看到完整版了,但是内容还是很有参考价值的。