安装SingleR包时 cannot open URL 'https://bioconductor.org/packages/3.16/workflows/bin/windows/contrib/4.2/PACKAGES'
时间: 2023-08-18 18:09:49 浏览: 411
这个错误通常是由于网络连接问题引起的。你可以尝试以下解决方案:
1. 确保你的网络连接正常,并且能够访问互联网。你可以尝试在浏览器中打开 https://bioconductor.org/packages/3.16/workflows/bin/windows/contrib/4.2/PACKAGES 确认是否能够成功打开。
2. 如果你使用的是公司或学校的网络,可能存在防火墙限制,导致无法访问某些网址。你可以尝试使用其他网络连接,如个人热点或其他网络环境。
3. 你可以尝试使用国内的镜像源进行安装。在安装前添加以下代码:
```r
options(BioC_mirror="https://mirrors.tuna.tsinghua.edu.cn/bioconductor/")
```
然后再尝试安装singleR:
```r
BiocManager::install("singleR")
```
这样可以使用清华大学的镜像源来下载所需的包。
如果以上方法都不奏效,可能是Bioconductor服务器暂时无法访问或存在其他问题。你可以稍后再尝试安装,或者到Bioconductor的官方网站上查找相关解决方案。希望能帮到你!
相关问题
cardiomyocyte.hesc <- SingleR(test = cardiomyocyteSingleR,ref = hpca.se, labels = hpca.se$label.main() + table(cardiomyocyte.hesc$labels,cardiomyocyte$seurat_clusters) Error: unexpected symbol in: "cardiomyocyte.hesc <- SingleR(test = cardiomyocyteSingleR,ref = hpca.se, labels = hpca.se$label.main() table
根据你提供的代码,你正在尝试使用SingleR函数进行单细胞RNA测序数据的比较,并使用table函数创建一个表格。然而,你在代码中出现了一个错误。在R中,函数调用通常不会在同一行中结束,除非你使用分号(;)来分隔多个语句。
以下是修正后的代码示例:
```R
cardiomyocyte.hesc <- SingleR(test = cardiomyocyteSingleR, ref = hpca.se, labels = hpca.se$label.main())
table(cardiomyocyte.hesc$labels, cardiomyocyte$seurat_clusters)
```
请确保在SingleR函数调用的末尾加上闭括号,并在table函数调用之前换行。
如果你还遇到其他问题,请提供更多的代码和错误信息,以便我能够更好地帮助你解决问题。
singleR怎么安装
要安装singleR,你需要遵循以下步骤:
1. 首先,你需要在R语言环境中安装包管理器"BiocManager"。如果你还没有安装它,可以使用以下命令安装:
```r
install.packages("BiocManager")
```
2. 安装singleR包:
```r
BiocManager::install("singleR")
```
3. 加载singleR包:
```r
library(singleR)
```
这样,你就可以开始使用singleR了。希望能帮助到你!
阅读全文
相关推荐
大家在看
以下为转载Plasma工作原理介紹-plasma等离子处理
以下为转载
Plasma工作原理介紹
工作原理
清洁效果的检验
Pull and Shear tests
Water contact angle measurement
Auger Electron Spectroscopic Analysis
Plasma机构原理圖
Plasma產生的原理
Plasma產生的條件
Ar/O2 Plasma的原理
Plasma Process
Plasma Parameter--(pc32系列)
Plasma 功效
早期,日本为了迎合高集成度的电子制造技术,开始使用超薄镀金技术,镀金厚度小于0.05mm。但问题也随之而来,当DM工艺后,经过烘烤,使原镀金层下的Ni元素会上移到表面。在随后的WB工艺中由于这些Ni元素及其他沾污会导致着线不佳现象,甚至着不上线(漏线,少线,第一点剥离,第二点剥离)。Plasma清洗机也就随之出现。
初版----劉卓 更新版----彭齊全
Oracle ASCP Profiles (Chinese version)
Oracle ASCP Profiles (Chinese version)
arcgis标准分幅图制作与生产
高速完成标准分幅图制作与生产等 高速完成
《程序设计基础》历年试题及答案.pdf
吉林大学计算机软件学院的历年期末试题,带答案的,可以参考,祝你高分
RealTek2797用户手册,最新
RealTek2797用户手册,最新的realtek芯片用户手册,支持2路HDMI和两路DP
最新推荐
036GraphTheory(图论) matlab代码.rar
1.版本:matlab2014/2019a/2024a
2.附赠案例数据可直接运行matlab程序。
3.代码特点:参数化编程、参数可方便更改、代码编程思路清晰、注释明细。
4.适用对象:计算机,电子信息工程、数学等专业的大学生课程设计、期末大作业和毕业设计。
026SVM用于分类时的参数优化,粒子群优化算法,用于优化核函数的c,g两个参数(SVM PSO)Matlab代码.rar
1.版本:matlab2014/2019a/2024a
2.附赠案例数据可直接运行matlab程序。
3.代码特点:参数化编程、参数可方便更改、代码编程思路清晰、注释明细。
4.适用对象:计算机,电子信息工程、数学等专业的大学生课程设计、期末大作业和毕业设计。
药店管理-JAVA-基于springBoot的药店管理系统的设计与实现(毕业论文+开题)
1. 用户角色
管理员
药店员工/药师
客户
2. 功能描述
管理员功能
用户管理
创建、编辑和删除药店员工和药师的账户。
设置不同用户的权限,确保敏感信息的安全。
库存管理
实时监控药品库存状态,设置库存预警,防止缺货或过期。
支持药品入库、出库和退货记录,自动更新库存数量。
商品管理
添加、编辑和删除药品信息,包括名称、规格、价格、生产厂家、有效期等。
分类管理药品,如处方药、非处方药、保健品等。
销售管理
查看和管理销售记录,生成每日、每周和每月的销售报表。
分析销售数据,了解畅销产品和季节性变化,以优化库存。
财务管理
监控药店的收入与支出,并生成财务报表。
管理支付方式(现金、信用卡、电子支付)及退款流程。
客户管理
记录客户的基本信息和购买历史,提供个性化服务。
管理会员制度,设置积分和优惠活动。
药品监管符合性
确保药店遵循相关法规,跟踪药品的进货渠道和销售记录。
提供合规报告,确保按规定进行药品管理。
报告与分析
生成各类统计报表,包括销售分析、库存分析和客户行为分析。
提供决策支持,帮助制定更好的经营策略。
药店员工/药师功能
销售操作
处理顾客的药
【网络】基于matlab高动态网络拓扑中OSPF网络计算【含Matlab源码 10964期】.zip
Matlab领域上传的视频是由对应的完整代码运行得来的,完整代码皆可运行,亲测可用,适合小白;
1、从视频里可见完整代码的内容
主函数:main.m;
调用函数:其他m文件;无需运行
运行结果效果图;
2、代码运行版本
Matlab 2019b;若运行有误,根据提示修改;若不会,私信博主;
3、运行操作步骤
步骤一:将所有文件放到Matlab的当前文件夹中;
步骤二:双击打开main.m文件;
步骤三:点击运行,等程序运行完得到结果;
4、仿真咨询
如需其他服务,可私信博主;
4.1 博客或资源的完整代码提供
4.2 期刊或参考文献复现
4.3 Matlab程序定制
4.4 科研合作
今天吴老师上课的时候说我.txt
今天吴老师上课的时候说我.txt
HTML挑战:30天技术学习之旅
资源摘要信息: "desafio-30dias"
标题 "desafio-30dias" 暗示这可能是一个与挑战或训练相关的项目,这在编程和学习新技能的上下文中相当常见。标题中的数字“30”很可能表明这个挑战涉及为期30天的时间框架。此外,由于标题是西班牙语,我们可以推测这个项目可能起源于或至少是针对西班牙语使用者的社区。标题本身没有透露技术上的具体内容,但挑战通常涉及一系列任务,旨在提升个人的某项技能或知识水平。
描述 "desafio-30dias" 并没有提供进一步的信息,它重复了标题的内容。因此,我们不能从中获得关于项目具体细节的额外信息。描述通常用于详细说明项目的性质、目标和期望成果,但由于这里没有具体描述,我们只能依靠标题和相关标签进行推测。
标签 "HTML" 表明这个挑战很可能与HTML(超文本标记语言)有关。HTML是构成网页和网页应用基础的标记语言,用于创建和定义内容的结构、格式和语义。由于标签指定了HTML,我们可以合理假设这个30天挑战的目的是学习或提升HTML技能。它可能包含创建网页、实现网页设计、理解HTML5的新特性等方面的任务。
压缩包子文件的文件名称列表 "desafio-30dias-master" 指向了一个可能包含挑战相关材料的压缩文件。文件名中的“master”表明这可能是一个主文件或包含最终版本材料的文件夹。通常,在版本控制系统如Git中,“master”分支代表项目的主分支,用于存放项目的稳定版本。考虑到这个文件名称的格式,它可能是一个包含所有相关文件和资源的ZIP或RAR压缩文件。
结合这些信息,我们可以推测,这个30天挑战可能涉及了一系列的编程任务和练习,旨在通过实践项目来提高对HTML的理解和应用能力。这些任务可能包括设计和开发静态和动态网页,学习如何使用HTML5增强网页的功能和用户体验,以及如何将HTML与CSS(层叠样式表)和JavaScript等其他技术结合,制作出丰富的交互式网站。
综上所述,这个项目可能是一个为期30天的HTML学习计划,设计给希望提升前端开发能力的开发者,尤其是那些对HTML基础和最新标准感兴趣的人。挑战可能包含了理论学习和实践练习,鼓励参与者通过构建实际项目来学习和巩固知识点。通过这样的学习过程,参与者可以提高在现代网页开发环境中的竞争力,为创建更加复杂和引人入胜的网页打下坚实的基础。
【CodeBlocks精通指南】:一步到位安装wxWidgets库(新手必备)

# 摘要
本文旨在为使用CodeBlocks和wxWidgets库的开发者提供详细的安装、配置、实践操作指南和性能优化建议。文章首先介绍了CodeBlocks和wxWidgets库的基本概念和安装流程,然后深入探讨了CodeBlocks的高级功能定制和wxWidgets的架构特性。随后,通过实践操作章节,指导读者如何创建和运行一个wxWidgets项目,包括界面设计、事件
andorid studio 配置ERROR: Cause: unable to find valid certification path to requested target
### 解决 Android Studio SSL 证书验证问题
当遇到 `unable to find valid certification path` 错误时,这通常意味着 Java 运行环境无法识别服务器提供的 SSL 证书。解决方案涉及更新本地的信任库或调整项目中的网络请求设置。
#### 方法一:安装自定义 CA 证书到 JDK 中
对于企业内部使用的私有 CA 颁发的证书,可以将其导入至 JRE 的信任库中:
1. 获取 `.crt` 或者 `.cer` 文件形式的企业根证书;
2. 使用命令行工具 keytool 将其加入 cacerts 文件内:
```
VC++实现文件顺序读写操作的技巧与实践
资源摘要信息:"vc++文件的顺序读写操作"
在计算机编程中,文件的顺序读写操作是最基础的操作之一,尤其在使用C++语言进行开发时,了解和掌握文件的顺序读写操作是十分重要的。在Microsoft的Visual C++(简称VC++)开发环境中,可以通过标准库中的文件操作函数来实现顺序读写功能。
### 文件顺序读写基础
顺序读写指的是从文件的开始处逐个读取或写入数据,直到文件结束。这与随机读写不同,后者可以任意位置读取或写入数据。顺序读写操作通常用于处理日志文件、文本文件等不需要频繁随机访问的文件。
### VC++中的文件流类
在VC++中,顺序读写操作主要使用的是C++标准库中的fstream类,包括ifstream(用于从文件中读取数据)和ofstream(用于向文件写入数据)两个类。这两个类都是从fstream类继承而来,提供了基本的文件操作功能。
### 实现文件顺序读写操作的步骤
1. **包含必要的头文件**:要进行文件操作,首先需要包含fstream头文件。
```cpp
#include <fstream>
```
2. **创建文件流对象**:创建ifstream或ofstream对象,用于打开文件。
```cpp
ifstream inFile("example.txt"); // 用于读操作
ofstream outFile("example.txt"); // 用于写操作
```
3. **打开文件**:使用文件流对象的成员函数open()来打开文件。如果不需要在创建对象时指定文件路径,也可以在对象创建后调用open()。
```cpp
inFile.open("example.txt", std::ios::in); // 以读模式打开
outFile.open("example.txt", std::ios::out); // 以写模式打开
```
4. **读写数据**:使用文件流对象的成员函数进行数据的读取或写入。对于读操作,可以使用 >> 运算符、get()、read()等方法;对于写操作,可以使用 << 运算符、write()等方法。
```cpp
// 读取操作示例
char c;
while (inFile >> c) {
// 处理读取的数据c
}
// 写入操作示例
const char *text = "Hello, World!";
outFile << text;
```
5. **关闭文件**:操作完成后,应关闭文件,释放资源。
```cpp
inFile.close();
outFile.close();
```
### 文件顺序读写的注意事项
- 在进行文件读写之前,需要确保文件确实存在,且程序有足够的权限对文件进行读写操作。
- 使用文件流进行读写时,应注意文件流的错误状态。例如,在读取完文件后,应检查文件流是否到达文件末尾(failbit)。
- 在写入文件时,如果目标文件不存在,某些open()操作会自动创建文件。如果文件已存在,open()操作则会清空原文件内容,除非使用了追加模式(std::ios::app)。
- 对于大文件的读写,应考虑内存使用情况,避免一次性读取过多数据导致内存溢出。
- 在程序结束前,应该关闭所有打开的文件流。虽然文件流对象的析构函数会自动关闭文件,但显式调用close()是一个好习惯。
### 常用的文件操作函数
- `open()`:打开文件。
- `close()`:关闭文件。
- `read()`:从文件读取数据到缓冲区。
- `write()`:向文件写入数据。
- `tellg()` 和 `tellp()`:分别返回当前读取位置和写入位置。
- `seekg()` 和 `seekp()`:设置文件流的位置。
### 总结
在VC++中实现顺序读写操作,是进行文件处理和数据持久化的基础。通过使用C++的标准库中的fstream类,我们可以方便地进行文件读写操作。掌握文件顺序读写不仅可以帮助我们在实际开发中处理数据文件,还可以加深我们对C++语言和文件I/O操作的理解。需要注意的是,在进行文件操作时,合理管理和异常处理是非常重要的,这有助于确保程序的健壮性和数据的安全。
【大数据时代必备:Hadoop框架深度解析】:掌握核心组件,开启数据科学之旅
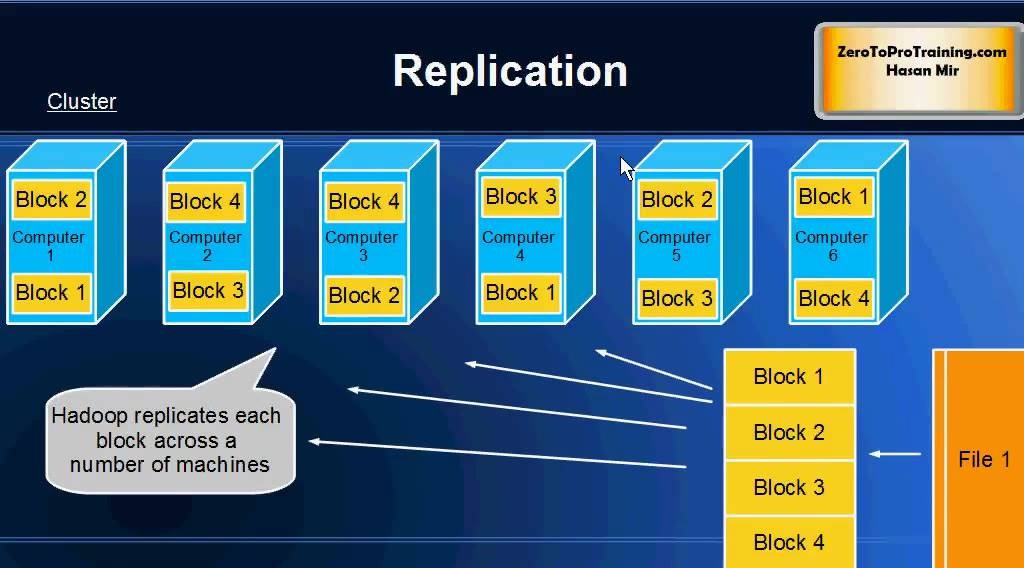
# 摘要
Hadoop作为一个开源的分布式存储和计算框架,在大数据处理领域发挥着举足轻重的作用。本文首先对Hadoop进行了概述,并介绍了其生态系统中的核心组件。深入分