Smith-Waterman算法
时间: 2024-05-21 14:16:47 浏览: 7
Smith-Waterman算法是用于比较两个序列的局部相似性的算法。它是一种动态规划算法,可以在比对DNA、RNA和蛋白质序列时被广泛使用。
该算法通过构建一个矩阵来比较两个序列,其中每个单元格表示两个序列中的一对位置的相似性得分。从这个矩阵中找到最高得分的单元格,然后通过回溯来确定匹配的位置。
具体来说,该算法首先创建一个矩阵,其中第i行和第j列的单元格表示第一个序列的前i个字符和第二个序列的前j个字符的相似性得分。然后,每个单元格的值都是由以下三个值中的最大值决定:
1. 左上角的单元格加上匹配得分(如果两个字符匹配)或惩罚得分(如果两个字符不匹配);
2. 向左移动的单元格加上惩罚得分;
3. 向上移动的单元格加上惩罚得分。
在计算完成后,最高得分的单元格就是最佳匹配的起点。从这个单元格开始,通过回溯来找到最佳匹配路径,直到到达得分为零的单元格为止。
Smith-Waterman算法在生物信息学和计算机科学中都有广泛的应用,例如在基因序列比对、蛋白质结构预测等领域。
相关问题
smith-waterman算法c++实现
Smith-Waterman算法是一种用于序列比对的动态规划算法。它可以用于比对DNA、RNA、蛋白质序列等。C++是一种高效的编程语言,可以用于实现Smith-Waterman算法。
实现Smith-Waterman算法的C++代码需要考虑以下几个方面:
1. 输入序列:需要从文件或者用户输入中读取待比对的序列。
2. 动态规划矩阵:需要创建一个二维数组来存储动态规划矩阵。
3. 算法实现:需要实现算法的核心部分,包括初始化矩阵、计算得分、回溯路径等。
4. 输出结果:需要将比对结果输出到文件或者屏幕上。
以下是一个简单的Smith-Waterman算法的C++实现示例:
```c++
#include <iostream>
#include <fstream>
#include <cstring>
using namespace std;
const int MAXLEN = 1000;
const int GAP = -2;
const int MATCH = 3;
const int MISMATCH = -1;
int score[MAXLEN][MAXLEN];
int max(int a, int b, int c) {
int m = a;
if (b > m) m = b;
if (c > m) m = c;
return m;
}
void init(int n, int m) {
for (int i = 0; i <= n; i++) {
score[i][0] = 0;
}
for (int j = 0; j <= m; j++) {
score[0][j] = 0;
}
}
void compute_score(char* s1, char* s2, int n, int m) {
for (int i = 1; i <= n; i++) {
for (int j = 1; j <= m; j++) {
int match = score[i-1][j-1] + (s1[i-1] == s2[j-1] ? MATCH : MISMATCH);
int delete_gap = score[i-1][j] + GAP;
int insert_gap = score[i][j-1] + GAP;
score[i][j] = max(match, delete_gap, insert_gap);
}
}
}
void traceback(char* s1, char* s2, int n, int m) {
int i = n, j = m;
while (i > 0 && j > 0) {
int match = score[i-1][j-1] + (s1[i-1] == s2[j-1] ? MATCH : MISMATCH);
int delete_gap = score[i-1][j] + GAP;
int insert_gap = score[i][j-1] + GAP;
if (score[i][j] == match) {
cout << s1[i-1] << " " << s2[j-1] << endl;
i--;
j--;
} else if (score[i][j] == delete_gap) {
cout << s1[i-1] << " -" << endl;
i--;
} else {
cout << "- " << s2[j-1] << endl;
j--;
}
}
}
int main() {
char s1[MAXLEN], s2[MAXLEN];
cin >> s1 >> s2;
int n = strlen(s1), m = strlen(s2);
init(n, m);
compute_score(s1, s2, n, m);
traceback(s1, s2, n, m);
return 0;
}
```
该示例代码实现了Smith-Waterman算法的核心部分,包括初始化矩阵、计算得分、回溯路径等。在输入两个待比对的序列后,程序会输出比对结果。需要注意的是,该示例代码只是一个简单的实现,实际应用中可能需要进行更多的优化和改进。
Use R,achieve Smith-Waterman
### 回答1:
Smith-Waterman算法是局部序列比对算法的一种,可以在R语言中通过以下代码实现:
首先,需要定义序列和得分矩阵:
```R
seq1 <- "ATCGGCTAGCG"
seq2 <- "TGGCGTA"
matchScore <- 2
mismatchScore <- -1
gapPenalty <- -2
```
接着,定义得分矩阵并初始化为0:
```R
n <- nchar(seq1)
m <- nchar(seq2)
scoreMatrix <- matrix(0, n+1, m+1)
```
然后,遍历得分矩阵,根据以下公式计算每个位置的得分:
```R
for (i in 2:(n+1)) {
for (j in 2:(m+1)) {
match <- scoreMatrix[i-1, j-1] + ifelse(substr(seq1, i-1, i-1) == substr(seq2, j-1, j-1), matchScore, mismatchScore)
delete <- scoreMatrix[i-1, j] + gapPenalty
insert <- scoreMatrix[i, j-1] + gapPenalty
scoreMatrix[i, j] <- max(0, match, delete, insert)
}
}
```
最后,在得分矩阵中找到得分最大的位置:
```R
maxScore <- max(scoreMatrix)
maxPos <- which(scoreMatrix == maxScore, arr.ind = TRUE)
endPos <- maxPos[1,]
```
根据得分矩阵和得分最大位置,可以重构局部相似区域的序列。
需要注意的是,该实现方式中,得分矩阵的第一行和第一列都初始化为0,因此,该算法只能找到得分非负的局部相似区域。如果需要找到得分负的局部相似区域,则需要对算法进行修改。
### 回答2:
Smith-Waterman算法是一种序列比对算法,在生物信息学中应用非常广泛。使用R语言可以实现Smith-Waterman算法,以下是实现的步骤:
1. 导入必要的R包。在R中,我们可以使用bioconductor包(如Biostrings或pairwiseAlignment函数)或者其他适用的包(如msa包)来处理序列对齐。
2. 准备待比对的序列。可以从文件中或者直接在代码中定义序列,格式可以是字符串或DNA/RNA序列对象。
3. 使用Smith-Waterman算法进行序列比对。根据上述步骤中导入的包的具体函数,调用相应的函数进行序列比对。Smith-Waterman算法会根据序列的相似性得分和gap惩罚,找到最佳的序列比对方式。
4. 解析比对结果。根据比对结果的格式,解析并提取所需信息,如得分、比对序列、比对位置等。这些信息可以进一步分析或可视化。
5. 进行后续分析。通过比对结果,可以进行多样性分析、进化分析、比对可视化等进一步的分析。根据具体需求,可以使用不同的R包和功能来完成进一步的分析。
以上是基本的步骤,当然具体实现过程可能因使用的R包和具体需求而有所不同。总之,使用R语言实现Smith-Waterman算法可以方便地进行序列比对,并且结合R中丰富的生物信息学工具和绘图功能,可以进行更深入的序列分析和可视化。
### 回答3:
使用R语言实现Smith-Waterman算法可以实现DNA序列对齐或蛋白质序列对齐等应用。下面是一个使用R语言实现Smith-Waterman算法的简单示例:
首先,我们需要定义一个函数来计算两个序列之间的得分矩阵。以下是一个示例函数,该函数计算两个序列之间的匹配得分矩阵:
```R
score_matrix <- function(seq1, seq2, match_score, mismatch_penalty, gap_penalty) {
m <- length(seq1)
n <- length(seq2)
score <- matrix(0, nrow = m + 1, ncol = n + 1)
for (i in 1:m) {
for (j in 1:n) {
match <- ifelse(seq1[i] == seq2[j], match_score, mismatch_penalty)
diagonal <- score[i, j] + match
up <- score[i + 1, j] + gap_penalty
left <- score[i, j + 1] + gap_penalty
score[i + 1, j + 1] <- max(diagonal, up, left, 0) # Smith-Waterman核心算法
}
}
return(score)
}
```
接下来,我们可以使用这个得分矩阵来回溯找到最优的序列对齐。以下是一个示例函数,该函数可以找到最优的序列对齐方式:
```R
align_sequences <- function(seq1, seq2, match_score, mismatch_penalty, gap_penalty) {
score <- score_matrix(seq1, seq2, match_score, mismatch_penalty, gap_penalty)
m <- length(seq1)
n <- length(seq2)
aligned_seq1 <- character()
aligned_seq2 <- character()
max_score <- max(score)
max_index <- which(score == max_score, arr.ind = TRUE)
start_row <- max_index[1, 1]
start_col <- max_index[1, 2]
i <- start_row
j <- start_col
while (score[i, j] != 0) {
if (score[i, j] == score[i - 1, j] + gap_penalty) {
aligned_seq1 <- c(seq1[i - 1], aligned_seq1)
aligned_seq2 <- c("-", aligned_seq2)
i <- i - 1
} else if (score[i, j] == score[i, j - 1] + gap_penalty) {
aligned_seq1 <- c("-", aligned_seq1)
aligned_seq2 <- c(seq2[j - 1], aligned_seq2)
j <- j - 1
} else {
aligned_seq1 <- c(seq1[i - 1], aligned_seq1)
aligned_seq2 <- c(seq2[j - 1], aligned_seq2)
i <- i - 1
j <- j - 1
}
}
alignment <- list(AlignmentScore = max_score, AlignedSeq1 = aligned_seq1, AlignedSeq2 = aligned_seq2)
return(alignment)
}
```
使用这两个函数,我们可以将两个序列进行对齐,得到最优的序列对齐结果。下面是一个示例:
```R
seq1 <- "ACGT"
seq2 <- "AAGGT"
match_score <- 1
mismatch_penalty <- -1
gap_penalty <- -2
alignment <- align_sequences(seq1, seq2, match_score, mismatch_penalty, gap_penalty)
```
以上就是使用R语言实现Smith-Waterman算法的简单示例。算法的核心思想是基于动态规划来寻找最优序列对齐方式,通过计算得分矩阵并进行回溯,可以得到最优的序列对齐结果。
相关推荐
最新推荐
序列比对-Needleman-Wunsch Smith-Waterman algorthm(java)
Needleman-Wunsch 算法(附java代码) Smith-Waterman 算法(附java代码)
基于springboot+vue+MySQL实现的在线考试系统+源代码+文档
web期末作业设计网页
基于springboot+vue+MySQL实现的在线考试系统+源代码+文档
318_面向物联网机器视觉的目标跟踪方法设计与实现的详细信息-源码.zip
提供的源码资源涵盖了安卓应用、小程序、Python应用和Java应用等多个领域,每个领域都包含了丰富的实例和项目。这些源码都是基于各自平台的最新技术和标准编写,确保了在对应环境下能够无缝运行。同时,源码中配备了详细的注释和文档,帮助用户快速理解代码结构和实现逻辑。
适用人群:
这些源码资源特别适合大学生群体。无论你是计算机相关专业的学生,还是对其他领域编程感兴趣的学生,这些资源都能为你提供宝贵的学习和实践机会。通过学习和运行这些源码,你可以掌握各平台开发的基础知识,提升编程能力和项目实战经验。
使用场景及目标:
在学习阶段,你可以利用这些源码资源进行课程实践、课外项目或毕业设计。通过分析和运行源码,你将深入了解各平台开发的技术细节和最佳实践,逐步培养起自己的项目开发和问题解决能力。此外,在求职或创业过程中,具备跨平台开发能力的大学生将更具竞争力。
其他说明:
为了确保源码资源的可运行性和易用性,特别注意了以下几点:首先,每份源码都提供了详细的运行环境和依赖说明,确保用户能够轻松搭建起开发环境;其次,源码中的注释和文档都非常完善,方便用户快速上手和理解代码;最后,我会定期更新这些源码资源,以适应各平台技术的最新发展和市场需求。
zigbee-cluster-library-specification
最新的zigbee-cluster-library-specification说明文档。
管理建模和仿真的文件
管理Boualem Benatallah引用此版本:布阿利姆·贝纳塔拉。管理建模和仿真。约瑟夫-傅立叶大学-格勒诺布尔第一大学,1996年。法语。NNT:电话:00345357HAL ID:电话:00345357https://theses.hal.science/tel-003453572008年12月9日提交HAL是一个多学科的开放存取档案馆,用于存放和传播科学研究论文,无论它们是否被公开。论文可以来自法国或国外的教学和研究机构,也可以来自公共或私人研究中心。L’archive ouverte pluridisciplinaire
MATLAB柱状图在信号处理中的应用:可视化信号特征和频谱分析
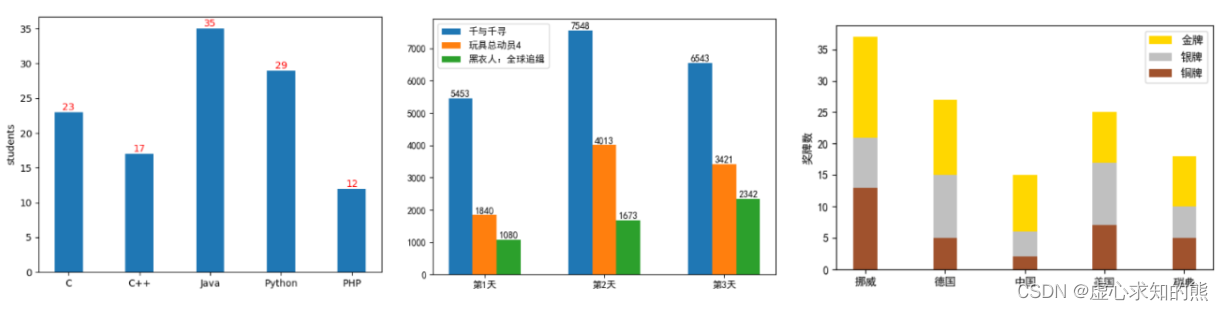
# 1. MATLAB柱状图概述**
MATLAB柱状图是一种图形化工具,用于可视化数据中不同类别或组的分布情况。它通过绘制垂直条形来表示每个类别或组中的数据值。柱状图在信号处理中广泛用于可视化信号特征和进行频谱分析。
柱状图的优点在于其简单易懂,能够直观地展示数据分布。在信号处理中,柱状图可以帮助工程师识别信号中的模式、趋势和异常情况,从而为信号分析和处理提供有价值的见解。
# 2. 柱状图在信号处理中的应用
柱状图在信号处理
hive中 的Metastore
Hive中的Metastore是一个关键的组件,它用于存储和管理Hive中的元数据。这些元数据包括表名、列名、表的数据类型、分区信息、表的存储位置等信息。Hive的查询和分析都需要Metastore来管理和访问这些元数据。
Metastore可以使用不同的后端存储来存储元数据,例如MySQL、PostgreSQL、Oracle等关系型数据库,或者Hadoop分布式文件系统中的HDFS。Metastore还提供了API,使得开发人员可以通过编程方式访问元数据。
Metastore的另一个重要功能是跟踪表的版本和历史。当用户对表进行更改时,Metastore会记录这些更改,并且可以让用户回滚到
JSBSim Reference Manual
JSBSim参考手册,其中包含JSBSim简介,JSBSim配置文件xml的编写语法,编程手册以及一些应用实例等。其中有部分内容还没有写完,估计有生之年很难看到完整版了,但是内容还是很有参考价值的。
"互动学习:行动中的多样性与论文攻读经历"
多样性她- 事实上SCI NCES你的时间表ECOLEDO C Tora SC和NCESPOUR l’Ingén学习互动,互动学习以行动为中心的强化学习学会互动,互动学习,以行动为中心的强化学习计算机科学博士论文于2021年9月28日在Villeneuve d'Asq公开支持马修·瑟林评审团主席法布里斯·勒菲弗尔阿维尼翁大学教授论文指导奥利维尔·皮耶昆谷歌研究教授:智囊团论文联合主任菲利普·普雷教授,大学。里尔/CRISTAL/因里亚报告员奥利维耶·西格德索邦大学报告员卢多维奇·德诺耶教授,Facebook /索邦大学审查员越南圣迈IMT Atlantic高级讲师邀请弗洛里安·斯特鲁布博士,Deepmind对于那些及时看到自己错误的人...3谢谢你首先,我要感谢我的两位博士生导师Olivier和Philippe。奥利维尔,"站在巨人的肩膀上"这句话对你来说完全有意义了。从科学上讲,你知道在这篇论文的(许多)错误中,你是我可以依
MATLAB柱状图在数据分析中的作用:从可视化到洞察
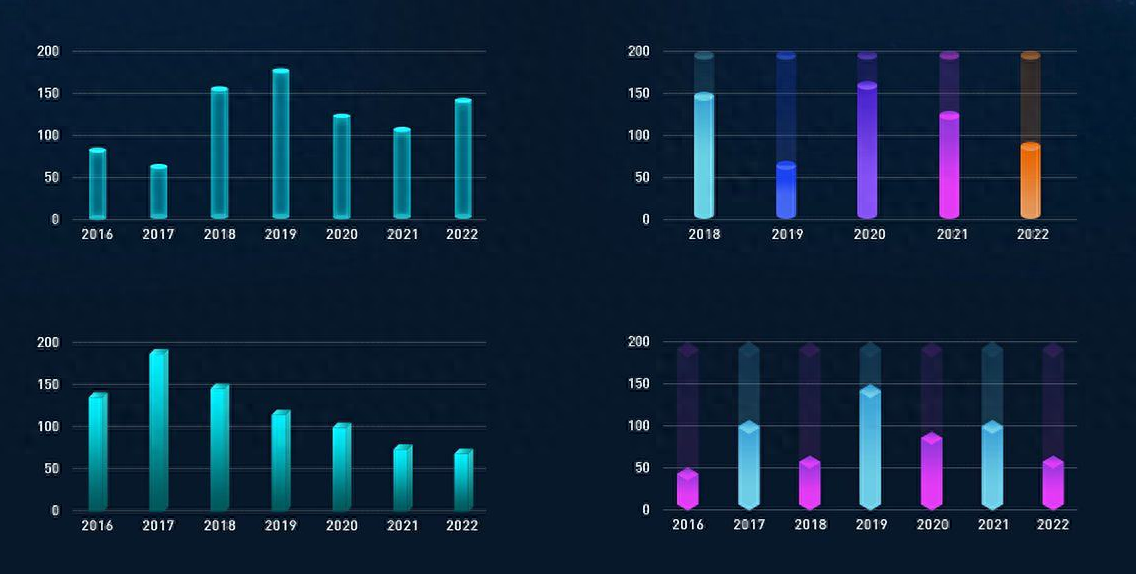
# 1. MATLAB柱状图概述**
柱状图是一种广泛用于数据可视化的图表类型,它使用垂直条形来表示数据中不同类别或组别的值。在MATLAB中,柱状图通过`bar`函数创建,该函数接受数据向量或矩阵作为输入,并生成相应的高度条形。
柱状图的优点在于其简单性和易于理解性。它们可以快速有效地传达数据分布和组别之间的比较。此外,MATLAB提供了广泛的定制选项,允许用户调整条形颜色、