cardiomyocyte <- RunPCA(cardiomyocyte, npcs=30,features = VariableFeatures(object = cardiomyocyte)) # Examine and visualize PCA results a few different ways
时间: 2024-04-07 19:30:31 浏览: 226
这段代码是用于对心肌细胞数据进行主成分分析(PCA)。它使用了"RunPCA"函数,并传入了参数npcs=30,表示要提取前30个主成分。另外,参数features=VariableFeatures(object = cardiomyocyte)表示只考虑具有变异性的特征进行PCA分析。
执行这段代码后,会得到PCA的结果,可以通过不同的方式进行查看和可视化。这可以帮助我们理解数据在主成分空间中的分布和结构。
相关问题
cardiomyocyteSingleR <- GetAssayData(cardiomyocyte,slot="data")# 获取标准化矩阵 cardiomyocyte.hesc <- SingleR(test = cardiomyocyteSingleR,ref = hpca.se, labels = hpca.se$label.main() cardiomyocyte.hesc # seurat 和 SingleR 的table表 table(cardiomyocyte.hesc$labels,cardiomyocyte$seurat_clusters) cardiomyocyte@meta.data$labels <- cardiomyocyte.hesc$labels
感谢提供代码!根据您提供的代码,您首先通过`GetAssayData`函数从`cardiomyocyte`对象中获取了标准化矩阵,并将其保存在`cardiomyocyteSingleR`变量中。
接下来,您使用了`SingleR`函数,将`cardiomyocyteSingleR`作为测试数据,`hpca.se`作为参考数据,并使用`hpca.se$label.main()`作为标签。结果保存在了`cardiomyocyte.hesc`变量中。
然后,您使用了`table`函数,以`cardiomyocyte.hesc$labels`和`cardiomyocyte$seurat_clusters`作为参数,生成了一个表格。
最后,您将`cardiomyocyte.hesc$labels`赋值给了`cardiomyocyte@meta.data$labels`。
请注意,代码中可能存在一些语法错误,我假设您已经处理过这些错误。如果您有任何更多的问题,请随时提问。
cardiomyocyte.hesc <- SingleR(test = cardiomyocyteSingleR,ref = hpca.se, labels = hpca.se$label.main() + table(cardiomyocyte.hesc$labels,cardiomyocyte$seurat_clusters) Error: unexpected symbol in: "cardiomyocyte.hesc <- SingleR(test = cardiomyocyteSingleR,ref = hpca.se, labels = hpca.se$label.main() table
根据你提供的代码,你正在尝试使用SingleR函数进行单细胞RNA测序数据的比较,并使用table函数创建一个表格。然而,你在代码中出现了一个错误。在R中,函数调用通常不会在同一行中结束,除非你使用分号(;)来分隔多个语句。
以下是修正后的代码示例:
```R
cardiomyocyte.hesc <- SingleR(test = cardiomyocyteSingleR, ref = hpca.se, labels = hpca.se$label.main())
table(cardiomyocyte.hesc$labels, cardiomyocyte$seurat_clusters)
```
请确保在SingleR函数调用的末尾加上闭括号,并在table函数调用之前换行。
如果你还遇到其他问题,请提供更多的代码和错误信息,以便我能够更好地帮助你解决问题。
阅读全文
相关推荐
大家在看
yolo开发人工智能小程序经验和总结.zip
yolo开发人工智能小程序经验和总结.zipyolo开发人工智能小程序经验和总结.zipyolo开发人工智能小程序经验和总结.zipyolo开发人工智能小程序经验和总结.zip
USB_HUB硬件电路引脚原理解析.docx
USB_HUB硬件电路引脚原理解析,与个人博文一致,这是word版本。
USB_HUB硬件电路引脚原理解析,与个人博文一致,这是word版本。
Keysight N6705C直流电源分析仪.pdf
Keysight N6705C直流电源分析仪
AS400 自学笔记集锦
AS400 自学笔记集锦
AS400学习笔记(V1.2)
自学使用的400操作命令集锦
LQR与PD控制在柔性机械臂中的对比研究
LQR与PD控制在柔性机械臂中的对比研究,路恩,杨雪锋,针对单杆柔性机械臂末端位置控制的问题,本文对柔性机械臂振动主动控制中较为常见的LQR和PD方法进行了控制效果的对比研究。首先,�
最新推荐
_三维电容层析成像组合电极激励测量模式.pdf
_三维电容层析成像组合电极激励测量模式
(1985-2024.6) 世界各国经济政策不确定性指数 (完整数据)
## 数据指标说明
经济政策不确定性(EPU)是指公众对未来政府经济政策方向、效果的不确定性。这种不确定性可能源于政治选举、政策变化预期、法规变动、国际关系紧张等多种因素。当政策不确定性较高时,企业和消费者可能会推迟投资和消费决策,从而影响经济活动
本次分享数据是全球及22个国家的经济政策不确定性指数,用于衡量各国经济政策的不确定性水平
数据介绍#井
数据名称:世界各国经济政策不确定性指数
数据年份:1985.1-2024.6
数据范围:22个国家
更新时间:2024年7月
数据来源:Economic Policy Uncertainty Index
数据说明:包括全球、中国、美国、日本等国家
数据用途##
用途:对全球格局、各国内部的经济政策不确定性进行量
从0开始C语言(1) C语言的数据类型
从0开始C语言(1)C语言的数据类型
课设项目Flask框架开发的微信公众号_订阅号消息自动回复服务.zip
1、该项目是团队成员近期最新开发,代码完整,资料齐全,含设计文档等,可直接用于毕设、课设、作业、比赛、学习
2、上传的项目源码经过严格测试,功能完善且能正常运行,请放心下载使用!
3、本项目适合计算机相关专业(人工智能、通信工程、自动化、电子信息、物联网等)的高校学生、教师、科研工作者、行业从业者下载使用,可借鉴学习,也可直接作为毕业设计、课程设计、作业、项目初期立项演示等,也适合小白学习进阶,遇到问题不懂就问,欢迎交流。
4、如果基础还行,可以在此代码基础上进行修改,以实现其他功能,也可直接用于毕设、课设、作业等。
5、不懂配置和运行,可远程教学
欢迎下载,学习使用!
提供项目开发过程中所有资料、很优质得项目。
精简博客系统-JAVA-基于Springboot开发的精简博客系统的设计与实现(毕业论文)
1. 用户管理
注册/登录: 用户通过邮箱或社交账号注册和登录。
用户角色: 支持不同角色(如管理员、作者、读者)管理权限。
2. 博客内容管理
文章发布: 用户可以创建、编辑和发布博客文章。
草稿保存: 支持将未完成的文章保存为草稿。
分类与标签: 提供对文章进行分类和添加标签的功能,便于整理内容。
3. 评论功能
评论系统: 读者可以对文章发表评论。
评论管理: 作者可管理评论,支持删除和审核功能。
4. 文章展示
首页展示: 以时间顺序或热度排序展示文章。
搜索功能: 提供关键词搜索功能,让用户快速找到文章。
5. 社交分享
分享链接: 一键生成文章分享链接,支持分享到各大社交平台。
6. 统计分析
访问统计: 提供文章阅读量及用户访问数据分析。
互动数据: 显示评论数和分享次数等互动指标。
7. 主题与个性化设置
主题选择: 提供多种主题样式供用户选择,以个性化博客外观。
自定义设置: 允许用户修改博客标题、简介及其他基本信息。
8. 安全与备份
数据安全: 定期自动备份文章和评论数据,防止丢失。
3dsmax高效建模插件Rappatools3.3发布,附教程
资源摘要信息:"Rappatools3.3.rar是一个与3dsmax软件相关的压缩文件包,包含了该软件的一个插件版本,名为Rappatools 3.3。3dsmax是Autodesk公司开发的一款专业的3D建模、动画和渲染软件,广泛应用于游戏开发、电影制作、建筑可视化和工业设计等领域。Rappatools作为一个插件,为3dsmax提供了额外的功能和工具,旨在提高用户的建模效率和质量。"
知识点详细说明如下:
1. 3dsmax介绍:
3dsmax,又称3D Studio Max,是一款功能强大的3D建模、动画和渲染软件。它支持多种工作流程,包括角色动画、粒子系统、环境效果、渲染等。3dsmax的用户界面灵活,拥有广泛的第三方插件生态系统,这使得它成为3D领域中的一个行业标准工具。
2. Rappatools插件功能:
Rappatools插件专门设计用来增强3dsmax在多边形建模方面的功能。多边形建模是3D建模中的一种技术,通过添加、移动、删除和修改多边形来创建三维模型。Rappatools提供了大量高效的工具和功能,能够帮助用户简化复杂的建模过程,提高模型的质量和完成速度。
3. 提升建模效率:
Rappatools插件中可能包含诸如自动网格平滑、网格优化、拓扑编辑、表面细分、UV展开等高级功能。这些功能可以减少用户进行重复性操作的时间,加快模型的迭代速度,让设计师有更多时间专注于创意和细节的完善。
4. 压缩文件内容解析:
本资源包是一个压缩文件,其中包含了安装和使用Rappatools插件所需的所有文件。具体文件内容包括:
- index.html:可能是插件的安装指南或用户手册,提供安装步骤和使用说明。
- license.txt:说明了Rappatools插件的使用许可信息,包括用户权利、限制和认证过程。
- img文件夹:包含用于文档或界面的图像资源。
- js文件夹:可能包含JavaScript文件,用于网页交互或安装程序。
- css文件夹:可能包含层叠样式表文件,用于定义网页或界面的样式。
5. MAX插件概念:
MAX插件指的是专为3dsmax设计的扩展软件包,它们可以扩展3dsmax的功能,为用户带来更多方便和高效的工作方式。Rappatools属于这类插件,通过在3dsmax软件内嵌入更多专业工具来提升工作效率。
6. Poly插件和3dmax的关系:
在3D建模领域,Poly(多边形)是构建3D模型的主要元素。所谓的Poly插件,就是指那些能够提供额外多边形建模工具和功能的插件。3dsmax本身就支持强大的多边形建模功能,而Poly插件进一步扩展了这些功能,为3dsmax用户提供了更多创建复杂模型的方法。
7. 增强插件的重要性:
在3D建模和设计行业中,增强插件对于提高工作效率和作品质量起着至关重要的作用。随着技术的不断发展和客户对视觉效果要求的提高,插件能够帮助设计师更快地完成项目,同时保持较高的创意和技术水准。
综上所述,Rappatools3.3.rar资源包对于3dsmax用户来说是一个很有价值的工具,它能够帮助用户在进行复杂的3D建模时提升效率并得到更好的模型质量。通过使用这个插件,用户可以在保持工作流程的一致性的同时,利用额外的工具集来优化他们的设计工作。
【R-Studio技术路径】:从RAID 5数据恢复基础到高级操作
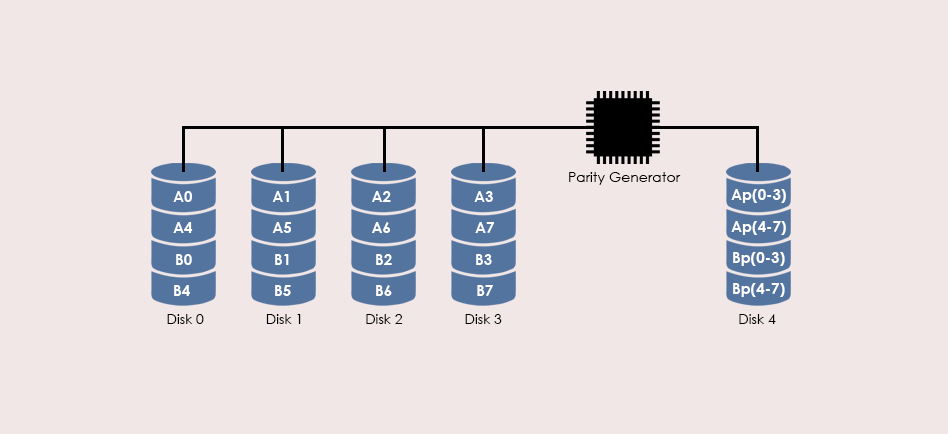
# 摘要
随着信息技术的发展,数据丢失问题日益突出,RAID 5作为常见的数据存储解决方案,其数据恢复技术显得尤为重要。本文首先介绍了RAID 5数据恢复的基础知识,然后详细解析了R-Studio软件的界面和核心功能,重点探讨了其在RAID 5数据恢复中的应用实践,包括磁盘镜像创建、数据提取、数据重组策略及一致性验证。进一步,本文还涉及了R-Studio的进阶技术,如脚本编
``` 定义1个圆类,成员有:1个半径成员变量,1个构造方法给成员变量赋初值,1个求面积方法。```定义1个圆类,成员有:1个半径成员变量,1个构造方法给成员变量赋初值,1个求面积方法。
当然,我们可以定义一个简单的`Circle`类,如下所示:
```java
public class Circle {
// 定义一个私有的半径成员变量
private double radius;
// 构造方法,用于初始化半径
public Circle(double initialRadius) {
this.radius = initialRadius;
}
// 求圆面积的方法
public double getArea() {
return Math.PI * Math.pow(radiu
Ruby实现PointInPolygon算法:判断点是否在多边形内
资源摘要信息:"PointInPolygon算法的Ruby实现是一个用于判断点是否在多边形内部的库。该算法通过计算点与多边形边界交叉线段的交叉次数来判断点是否在多边形内部。如果交叉数为奇数,则点在多边形内部,如果为偶数或零,则点在多边形外部。库中包含Pinp::Point类和Pinp::Polygon类。Pinp::Point类用于表示点,Pinp::Polygon类用于表示多边形。用户可以向Pinp::Polygon中添加点来构造多边形,然后使用contains_point?方法来判断任意一个Pinp::Point对象是否在该多边形内部。"
1. Ruby语言基础:Ruby是一种动态、反射、面向对象、解释型的编程语言。它具有简洁、灵活的语法,使得编写程序变得简单高效。Ruby语言广泛用于Web开发,尤其是Ruby on Rails这一著名的Web开发框架就是基于Ruby语言构建的。
2. 类和对象:在Ruby中,一切皆对象,所有对象都属于某个类,类是对象的蓝图。Ruby支持面向对象编程范式,允许程序设计者定义类以及对象的创建和使用。
3. 算法实现细节:算法基于数学原理,即计算点与多边形边界线段的交叉次数。当点位于多边形内时,从该点出发绘制射线与多边形边界相交的次数为奇数;如果点在多边形外,交叉次数为偶数或零。
4. Pinp::Point类:这是一个表示二维空间中的点的类。类的实例化需要提供两个参数,通常是点的x和y坐标。
5. Pinp::Polygon类:这是一个表示多边形的类,由若干个Pinp::Point类的实例构成。可以使用points方法添加点到多边形中。
6. contains_point?方法:属于Pinp::Polygon类的一个方法,它接受一个Pinp::Point类的实例作为参数,返回一个布尔值,表示传入的点是否在多边形内部。
7. 模块和命名空间:在Ruby中,Pinp是一个模块,模块可以用来将代码组织到不同的命名空间中,从而避免变量名和方法名冲突。
8. 程序示例和测试:Ruby程序通常包含方法调用、实例化对象等操作。示例代码提供了如何使用PointInPolygon算法进行点包含性测试的基本用法。
9. 边缘情况处理:算法描述中提到要添加选项测试点是否位于多边形的任何边缘。这表明算法可能需要处理点恰好位于多边形边界的情况,这类点在数学上可以被认为是既在多边形内部,又在多边形外部。
10. 文件结构和工程管理:提供的信息表明有一个名为"PointInPolygon-master"的压缩包文件,表明这可能是GitHub等平台上的一个开源项目仓库,用于管理PointInPolygon算法的Ruby实现代码。文件名称通常反映了项目的版本管理,"master"通常指的是项目的主分支,代表稳定版本。
11. 扩展和维护:算法库像PointInPolygon这类可能需要不断维护和扩展以适应新的需求或修复发现的错误。开发者会根据实际应用场景不断优化算法,同时也会有社区贡献者参与改进。
12. 社区和开源:Ruby的开源生态非常丰富,Ruby开发者社区非常活跃。开源项目像PointInPolygon这样的算法库在社区中广泛被使用和分享,这促进了知识的传播和代码质量的提高。
以上内容是对给定文件信息中提及的知识点的详细说明。根据描述,该算法库可用于各种需要点定位和多边形空间分析的场景,例如地理信息系统(GIS)、图形用户界面(GUI)交互、游戏开发、计算机图形学等领域。
【R-Studio恢复工具解析】:RAID 5恢复的功能优势与实际应用

# 摘要
RAID 5技术因其高效的数据存储和容错能力被广泛应用。然而,数据丢失问题仍时有发生,R-Studio作为一种功能强大的恢复工具,为解决这一问题提供了有效的技术方案。本文概述了RAID 5的基本概念、R-Studio的理论基础及其数据恢复原理。通过分析R-Studio的主要功能和恢复流程,本文还探讨了该工具