给我选择一个数据库进行生物信息学的完整分析,包括一系列的分析流程以及代码,等等
时间: 2024-11-15 07:32:31 浏览: 2

Bioinformatics生物信息学:序列和基因组分析.pdf
选择一个适合生物信息学完整分析的数据库会因项目需求而异,但以Ensembl为例,它是一个常用的生物信息学资源库,涵盖了基因组、转录组、蛋白质等多个层面。下面是使用Ensembl进行完整分析的一个基本步骤,并简要提到了相关的Python代码片段:
1. **获取数据**
使用Ensembl提供的REST API 或 BioPython 的 `Ensembl RESTful Client` 来获取基因组、转录本、蛋白质等数据。例如,下载特定物种的基因组FASTA文件:
```python
from Bio import EnsemblRest
ensembl_client = EnsemblRest.EnsemblClient('ensembl.org')
fasta_url = ensembl_client.get_fasta_sequence('Homo sapiens', 'GRCh38', 'chr1')
```
2. **序列分析**
对下载的序列数据进行比对、拼接、剪切等操作。例如,使用BLAST查找相似序列:
```python
from Bio.Blast import NCBIWWW
result_handle = NCBIWWW.qblast("blastn", "nt", sequence)
blast_record = Blast.Record(result_handle)
```
3. **基因注释**
使用Ensembl提供的基因注解工具(如Exonerate或Sailfish),将测序数据与基因模型比对:
```python
exonerate_results = exonerate.sequence_to_gff3(sequence, ensembl_genome)
```
4. **转录组分析**
利用RNA-seq数据处理工具(如Hisat2、StringTie或DESeq2)进行转录本组装和差异表达分析:
```python
!hisat2 -x {ensembl_index} -U reads.fastq.gz | stringtie --threads 4 -o transcripts.gtf
```
分析结果通常导出为表格形式进行统计和绘图:
```python
!deseq2 -r transcripts.gtf > results.csv
```
5. **蛋白质功能分析**
对编码蛋白质的特征进行分析,比如计算功能域、预测蛋白质结构和进行蛋白质互作网络构建:
```python
# 使用InterProScan
!interproscan.sh -i protein.fasta -f tsv -d ipr -out interpro.tsv
```
6. **可视化**
结果可视化工具如R的ggplot2、Bioconductor的ComplexHeatmap等可以展示基因表达热图、网络图等:
```python
import matplotlib.pyplot as plt
df.plot(kind='bar', figsize=(10, 5))
plt.show()
```
这个过程只是一个基本框架,实际分析中可能需要结合其他工具和技术,如生物信息学软件如BAMTools、Bedtools、BedGraphToBigWig等。具体的代码和分析流程需根据项目的具体需求和数据类型进行调整。
阅读全文
相关推荐
最新推荐
完美解决SQL server 5173问题(一个或多个文件与数据库的主文件不匹配)
在SQL Server中,当尝试附加一个数据库时,可能会遇到错误5173,这通常意味着一个或多个文件与数据库的主文件不匹配。错误信息表明数据库的MDF(主数据文件)和LDF(日志文件)之间存在不一致性,可能是由于文件损坏...
数据库选型分析.docx
对于需要高度可扩展性和容错性的企业,Datastax可能是一个不错的选择。 除了图数据库,其他类型的数据库如Oracle(关系型数据库)、Redis(键值存储)、MongoDB(文档数据库)和Elasticsearch(搜索引擎)都是各自...
利用Rational_Rose进行C++代码和数据库结构分析.doc
Rational Rose是一款强大的UML建模工具,特别适用于C++代码和数据库结构的分析。它支持UML(统一建模语言)的各个模型图,包括类图、数据模型图和组件图,帮助开发者理解现有系统的架构。逆向工程是Rational Rose的...
DB2数据库网络协议分析报告
每个结构都包含一个DDM头,包括长度字段、魔数(Magic)、格式标识和相关ID(CorrelID)。报文的后续部分包含代码点(Code point),用于指明数据的用途,以及数据区,数据区可能是参数结构或直接的字节流。参数结构...
MySQL 修改数据库名称的一个新奇方法
我们利用`INFORMATION_SCHEMA.TABLES`视图来获取旧数据库中所有表的信息,然后构建一个SQL脚本,将每个表从旧数据库迁移到新数据库: ```sql mysql> SELECT CONCAT("RENAME TABLE ", table_schema, ".", table_name...
平尾装配工作平台运输支撑系统设计与应用
资源摘要信息:"该压缩包文件名为‘行业分类-设备装置-用于平尾装配工作平台的运输支撑系统.zip’,虽然没有提供具体的标签信息,但通过文件标题可以推断出其内容涉及的是航空或者相关重工业领域内的设备装置。从标题来看,该文件集中讲述的是有关平尾装配工作平台的运输支撑系统,这是一种专门用于支撑和运输飞机平尾装配的特殊设备。
平尾,即水平尾翼,是飞机尾部的一个关键部件,它对于飞机的稳定性和控制性起到至关重要的作用。平尾的装配工作通常需要在一个特定的平台上进行,这个平台不仅要保证装配过程中平尾的稳定,还需要适应平尾的搬运和运输。因此,设计出一个合适的运输支撑系统对于提高装配效率和保障装配质量至关重要。
从‘用于平尾装配工作平台的运输支撑系统.pdf’这一文件名称可以推断,该PDF文档应该是详细介绍这种支撑系统的构造、工作原理、使用方法以及其在平尾装配工作中的应用。文档可能包括以下内容:
1. 支撑系统的设计理念:介绍支撑系统设计的基本出发点,如便于操作、稳定性高、强度大、适应性强等。可能涉及的工程学原理、材料学选择和整体结构布局等内容。
2. 结构组件介绍:详细介绍支撑系统的各个组成部分,包括支撑框架、稳定装置、传动机构、导向装置、固定装置等。对于每一个部件的功能、材料构成、制造工艺、耐腐蚀性以及与其他部件的连接方式等都会有详细的描述。
3. 工作原理和操作流程:解释运输支撑系统是如何在装配过程中起到支撑作用的,包括如何调整支撑点以适应不同重量和尺寸的平尾,以及如何进行运输和对接。操作流程部分可能会包含操作步骤、安全措施、维护保养等。
4. 应用案例分析:可能包含实际操作中遇到的问题和解决方案,或是对不同机型平尾装配过程的支撑系统应用案例的详细描述,以此展示系统的实用性和适应性。
5. 技术参数和性能指标:列出支撑系统的具体技术参数,如载重能力、尺寸规格、工作范围、可调节范围、耐用性和可靠性指标等,以供参考和评估。
6. 安全和维护指南:对于支撑系统的使用安全提供指导,包括操作安全、应急处理、日常维护、定期检查和故障排除等内容。
该支撑系统作为专门针对平尾装配而设计的设备,对于飞机制造企业来说,掌握其详细信息是提高生产效率和保障产品质量的重要一环。同时,这种支撑系统的设计和应用也体现了现代工业在专用设备制造方面追求高效、安全和精确的趋势。"
管理建模和仿真的文件
管理Boualem Benatallah引用此版本:布阿利姆·贝纳塔拉。管理建模和仿真。约瑟夫-傅立叶大学-格勒诺布尔第一大学,1996年。法语。NNT:电话:00345357HAL ID:电话:00345357https://theses.hal.science/tel-003453572008年12月9日提交HAL是一个多学科的开放存取档案馆,用于存放和传播科学研究论文,无论它们是否被公开。论文可以来自法国或国外的教学和研究机构,也可以来自公共或私人研究中心。L’archive ouverte pluridisciplinaire
MATLAB遗传算法探索:寻找随机性与确定性的平衡艺术
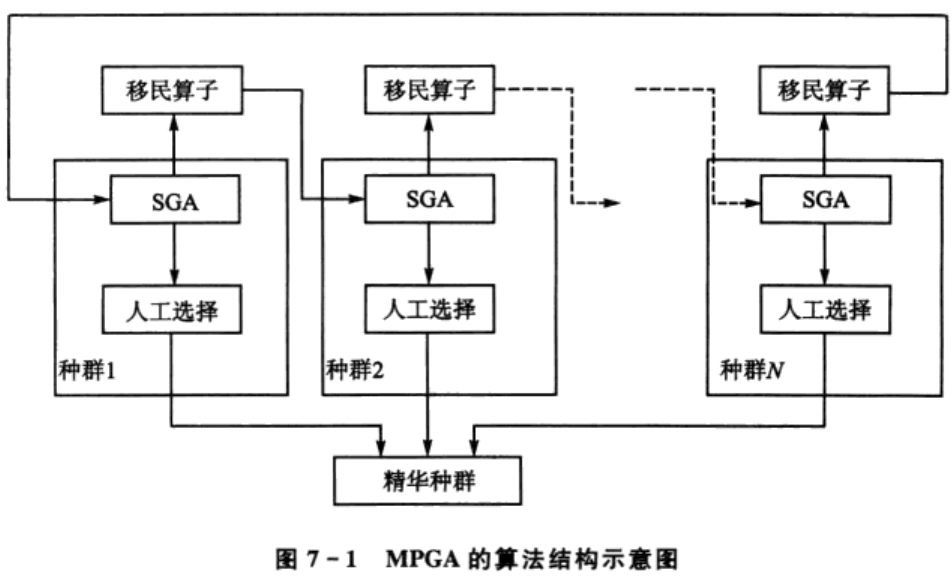
# 1. 遗传算法的基本概念与起源
遗传算法(Genetic Algorithm, GA)是一种模拟自然选择和遗传学机制的搜索优化算法。起源于20世纪60年代末至70年代初,由John Holland及其学生和同事们在研究自适应系统时首次提出,其理论基础受到生物进化论的启发。遗传算法通过编码一个潜在解决方案的“基因”,构造初始种群,并通过选择、交叉(杂交)和变异等操作模拟生物进化过程,以迭代的方式不断优化和筛选出最适应环境的
如何在S7-200 SMART PLC中使用MB_Client指令实现Modbus TCP通信?请详细解释从连接建立到数据交换的完整步骤。
为了有效地掌握S7-200 SMART PLC中的MB_Client指令,以便实现Modbus TCP通信,建议参考《S7-200 SMART Modbus TCP教程:MB_Client指令与功能码详解》。本教程将引导您了解从连接建立到数据交换的整个过程,并详细解释每个步骤中的关键点。
参考资源链接:[S7-200 SMART Modbus TCP教程:MB_Client指令与功能码详解](https://wenku.csdn.net/doc/119yes2jcm?spm=1055.2569.3001.10343)
首先,确保您的S7-200 SMART CPU支持开放式用户通
MAX-MIN Ant System:用MATLAB解决旅行商问题
资源摘要信息:"Solve TSP by MMAS: Using MAX-MIN Ant System to solve Traveling Salesman Problem - matlab开发"
本资源为解决经典的旅行商问题(Traveling Salesman Problem, TSP)提供了一种基于蚁群算法(Ant Colony Optimization, ACO)的MAX-MIN蚁群系统(MAX-MIN Ant System, MMAS)的Matlab实现。旅行商问题是一个典型的优化问题,要求找到一条最短的路径,让旅行商访问每一个城市一次并返回起点。这个问题属于NP-hard问题,随着城市数量的增加,寻找最优解的难度急剧增加。
MAX-MIN Ant System是一种改进的蚁群优化算法,它在基本的蚁群算法的基础上,对信息素的更新规则进行了改进,以期避免过早收敛和局部最优的问题。MMAS算法通过限制信息素的上下界来确保算法的探索能力和避免过早收敛,它在某些情况下比经典的蚁群系统(Ant System, AS)和带有局部搜索的蚁群系统(Ant Colony System, ACS)更为有效。
在本Matlab实现中,用户可以通过调用ACO函数并传入一个TSP问题文件(例如"filename.tsp")来运行MMAS算法。该问题文件可以是任意的对称或非对称TSP实例,用户可以从特定的网站下载多种标准TSP问题实例,以供测试和研究使用。
使用此资源的用户需要注意,虽然该Matlab代码可以免费用于个人学习和研究目的,但若要用于商业用途,则需要联系作者获取相应的许可。作者的电子邮件地址为***。
此外,压缩包文件名为"MAX-MIN%20Ant%20System.zip",该压缩包包含Matlab代码文件和可能的示例数据文件。用户在使用之前需要将压缩包解压,并将文件放置在Matlab的适当工作目录中。
为了更好地理解和应用该资源,用户应当对蚁群优化算法有初步了解,尤其是对MAX-MIN蚁群系统的基本原理和运行机制有所掌握。此外,熟悉Matlab编程环境和拥有一定的编程经验将有助于用户根据个人需求修改和扩展算法。
在实际应用中,用户可以根据问题规模调整MMAS算法的参数,如蚂蚁数量、信息素蒸发率、信息素增量等,以获得最优的求解效果。此外,也可以结合其他启发式或元启发式算法,如遗传算法、模拟退火等,来进一步提高算法的性能。
总之,本资源为TSP问题的求解提供了一种有效的算法框架,且Matlab作为编程工具的易用性和强大的计算能力,使得该资源成为算法研究人员和工程技术人员的有力工具。通过本资源的应用,用户将能够深入探索并实现蚁群优化算法在实际问题中的应用,为解决复杂的优化问题提供一种新的思路和方法。