有一个vcf文件,在xshell服务器使用vcfrools提取2号染色体的SNP位点,在Haploview进行单倍型分析的具体步骤和代码
时间: 2024-12-22 17:31:21 浏览: 35
在XShell服务器上使用VCFtools提取2号染色体的SNP位点,并在Haploview进行单倍型分析的过程需要分几个步骤:
1. **安装必要的工具**:
- VCFtools:如果你还没有安装,可以访问[这里](https://vcftools.github.io/)下载并按照说明安装。
- Haploview:这是一个专门用于查看遗传标记单倍型数据的软件,可以在Haploview官网下载并安装。
2. **打开VCF文件**:
使用VCFtools的`vcf-query`命令定位到含有2号染色体信息的部分。假设你的VCF文件名为`example.vcf`:
```
vcf-query example.vcf -chr 2 > chromosome_2_snps.vcf
```
这将创建一个新的VCF文件,仅包含2号染色体的SNP。
3. **过滤SNPs**:
确保只选择你感兴趣的SNP位点,比如那些有足够样本覆盖、低缺失率等。你可以用`vcf-filter`命令添加条件:
```
vcf-filter chromosome_2_snps.vcf -maf 0.05 > filtered_snps.vcf
```
4. **导入Haploview**:
下载并解压Haploview安装包后,打开Haploview软件。从菜单中选择“File”>“Load Variant Data”,加载`filtered_snps.vcf`文件。
5. **单倍型分析**:
- 创建一个项目:点击“Create New Project”。
- 导入数据:将刚刚加载的SNP数据导入项目。
- 分析:选择“View”>“Single Sample View”或“Multi-sample View”,然后可以选择不同的分析选项,如查看单体型、LD图等。
6. **保存结果**:
完成分析后,可以导出图片或者其他格式的结果以便后续研究。
**注意:**以上操作可能会因具体的系统设置和软件版本有所不同。在实际操作前,确保你已经对这两个工具的操作有所了解。
阅读全文
相关推荐
大家在看
LTE Signaling & Protocol Analysis Focus: E-UTRAN and UE
非常不错,采用问答的方式来学习LTE和EPC,本章主要关注于UE和RAN部分。
This eBook is a must for everybody who requires a detailed understanding of the protocols and signaling procedures within E-UTRAN and the EPC. In that respect the clear focus of this course is on the protocols of the UE and the E-UTRAN.
The eBook starts with a review of the LTE physical layer and the concepts and protocol stacks of E-UTRAN.
This part concludes with the review of the EPS network architecture.
Immediately afterwards we jump into real-life call flows and scenarios and confront the student with the look & feel of the LTE protocol suite. This part ends with an assessment of what will be the focus of the following chapters.
The next chapters are dedicated to the different protocols EMM, ESM, MAC, RLC, RRC, S1-AP, X2-AP, SGs-AP and S101-AP.
The eBook concludes with the presentation and analysis of LTE signaling flows and real-life call flows.
任务执行器-用于ad9834波形发生器(dds)的幅度控制电路
7.2 任务执行器
堆垛机
概述
堆垛机是一种特殊类型的运输机,专门设计用来与货架一起工作。堆垛机在两排货架间的巷
道中往复滑行,提取和存入临时实体。堆垛机可以充分展示伸叉、提升和行进动作。提升和
行进运动是同时进行的,但堆垛机完全停车后才会进行伸叉。
详细说明
堆垛机是任务执行器的一个子类。它通过沿着自身x轴方向行进的方式来实现偏移行进。它
一直行进直到与目的地位置正交,并抬升其载货平台。如果偏移行进是要执行装载或卸载任
务,那么一完成偏移,它就会执行用户定义的装载/卸载时间,将临时实体搬运到其载货平
台,或者从其载货平台搬运到目的位置。
默认情况下,堆垛机不与导航器相连。这意味着不执行行进任务。取尔代之,所有行进都采
用偏移行进的方式完成。
关于将临时实体搬运到堆垛机上的注释:对于一个装载任务,如果临时实体处于一个不断刷
新临时实体位置的实体中,如传送带时,堆垛机就不能将临时实体搬运到载货平台上。这种
情况下,如果想要显示将临时实体搬运到载货平台的过程,则需确保在模型树中,堆垛机排
在它要提取临时实体的那个实体的后面(在模型树中,堆垛机必须排在此实体下面)。
除了任务执行器所具有的标准属性外,堆垛机具有建模人员定义的载货平台提升速度和初始
提升位置。当堆垛机空闲或者没有执行偏移行进任务时,载货平台将回到此初始位置的高度。
332
美国Flexsim公司&北京创时能科技发展有限公司版权所有【010-82780244】
不同拉压模量弹性力学问题研究的新进展
不同拉压模量弹性力学问题研究的新进展,赵慧玲,叶志明,拉压不同模量弹性体具有材料非线性特征,不同模量本构关系受到材料本身及结构各点的应力、应变状态等因素的综合影响。本文总结了
【管道瞬变流】特征线法管道瞬变流计算【含Matlab源码 2773期】.zip
Matlab领域上传的全部代码均可运行,亲测可用,尽我所能,为你服务;
1、代码压缩包内容
主函数:main.m;
调用函数:其他m文件;无需运行
运行结果效果图;
2、代码运行版本
Matlab 2019b;若运行有误,根据提示修改;若不会,可私信博主;
3、运行操作步骤
步骤一:将所有文件放到Matlab的当前文件夹中;
步骤二:双击打开main.m文件;
步骤三:点击运行,等程序运行完得到结果;
4、物理应用
仿真:导航、地震、电磁、电路、电能、机械、工业控制、水位控制、直流电机、平面电磁波、管道瞬变流、刚度计算
光学:光栅、杨氏双缝、单缝、多缝、圆孔、矩孔衍射、夫琅禾费、干涉、拉盖尔高斯、光束、光波、涡旋
定位问题:chan、taylor、RSSI、music、卡尔曼滤波UWB
气动学:弹道、气体扩散、龙格库弹道
运动学:倒立摆、泊车
天体学:卫星轨道、姿态
船舶:控制、运动
电磁学:电场分布、电偶极子、永磁同步、变压器
天线测试手册
能不说么?实在是没说的了。其实就这点了,真的,实在没说的了
最新推荐
科研工作量管理系统(代码+数据库+LW)
摘 要
现代经济快节奏发展以及不断完善升级的信息化技术,让传统数据信息的管理升级为软件存储,归纳,集中处理数据信息的管理方式。本科研工作量管理系统就是在这样的大环境下诞生,其可以帮助管理者在短时间内处理完毕庞大的数据信息,使用这种软件工具可以帮助管理人员提高事务处理效率,达到事半功倍的效果。此科研工作量管理系统利用当下成熟完善的SSM框架,使用跨平台的可开发大型商业网站的Java语言,以及最受欢迎的RDBMS应用软件之一的Mysql数据库进行程序开发。实现了用户在线选择试题并完成答题,在线查看考核分数。管理员管理字典管理、工作量管理、科研获奖管理、科研论文管理、秘书管理、科研项目管理、教师管理、管理员管理等功能。科研工作量管理系统的开发根据操作人员需要设计的界面简洁美观,在功能模块布局上跟同类型网站保持一致,程序在实现基本要求功能时,也为数据信息面临的安全问题提供了一些实用的解决方案。可以说该程序在帮助管理者高效率地处理工作事务的同时,也实现了数据信息的整体化,规范化与自动化。
关键词:科研工作量管理系统;SSM框架;Mysql;自动化
租赁合同编写指南及下载资源
资源摘要信息:《租赁合同》是用于明确出租方与承租方之间的权利和义务关系的法律文件。在实际操作中,一份详尽的租赁合同对于保障交易双方的权益至关重要。租赁合同应当包括但不限于以下要点:
1. 双方基本信息:租赁合同中应明确出租方(房东)和承租方(租客)的名称、地址、联系方式等基本信息。这对于日后可能出现的联系、通知或法律诉讼具有重要意义。
2. 房屋信息:合同中需要详细说明所租赁的房屋的具体信息,包括房屋的位置、面积、结构、用途、设备和家具清单等。这些信息有助于双方对租赁物有清晰的认识。
3. 租赁期限:合同应明确租赁开始和结束的日期,以及租期的长短。租赁期限的约定关系到租金的支付和合同的终止条件。
4. 租金和押金:租金条款应包括租金金额、支付周期、支付方式及押金的数额。同时,应明确规定逾期支付租金的处理方式,以及押金的退还条件和时间。
5. 维修与保养:在租赁期间,房屋的维护和保养责任应明确划分。通常情况下,房东负责房屋的结构和主要设施维修,而租客需负责日常维护及保持房屋的清洁。
6. 使用与限制:合同应规定承租方可以如何使用房屋以及可能的限制。例如,禁止非法用途、允许或禁止宠物、是否可以转租等。
7. 终止与续租:租赁合同应包括租赁关系的解除条件,如提前通知时间、违约责任等。同时,双方可以在合同中约定是否可以续租,以及续租的条件。
8. 解决争议的条款:合同中应明确解决可能出现的争议的途径,包括适用法律、管辖法院等,有助于日后纠纷的快速解决。
9. 其他可能需要的条款:根据具体情况,合同中可能还需要包括关于房屋保险、税费承担、合同变更等内容。
下载资源链接:【下载自www.glzy8.com管理资源吧】Rental contract.DOC
该资源为一份租赁合同模板,对需要进行房屋租赁的个人或机构提供了参考价值。通过对合同条款的详细列举和解释,该文档有助于用户了解和制定自己的租赁合同,从而在房屋租赁交易中更好地保护自己的权益。感兴趣的用户可以通过提供的链接下载文档以获得更深入的了解和实际操作指导。
【项目管理精英必备】:信息系统项目管理师教程习题深度解析(第四版官方教材全面攻略)
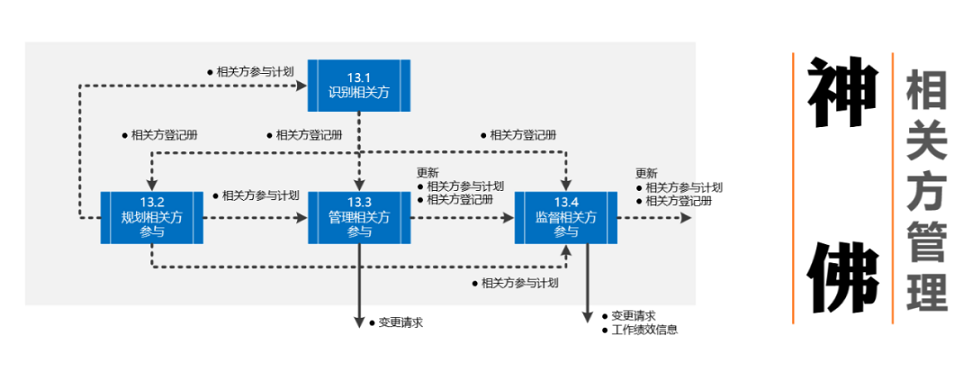
# 摘要
信息系统项目管理是确保项目成功交付的关键活动,涉及一系列管理过程和知识领域。本文深入探讨了信息系统项目管理的各个方面,包括项目管理过程组、知识领域、实践案例、管理工具与技术,以及沟通和团队协作。通过分析不同的项目管理方法论(如瀑布、迭代、敏捷和混合模型),并结合具体案例,文章阐述了项目管理的最佳实践和策略。此外,本文还涵盖了项目管理中的沟通管理、团队协作的重要性,
最具代表性的改进过的UNet有哪些?
UNet是一种广泛用于图像分割任务的卷积神经网络结构,它的特点是结合了下采样(编码器部分)和上采样(解码器部分),能够保留细节并生成精确的边界。为了提高性能和适应特定领域的需求,研究者们对原始UNet做了许多改进,以下是几个最具代表性的变种:
1. **DeepLab**系列:由Google开发,通过引入空洞卷积(Atrous Convolution)、全局平均池化(Global Average Pooling)等技术,显著提升了分辨率并保持了特征的多样性。
2. **SegNet**:采用反向传播的方式生成全尺寸的预测图,通过上下采样过程实现了高效的像素级定位。
3. **U-Net+
惠普P1020Plus驱动下载:办公打印新选择
资源摘要信息: "最新惠普P1020Plus官方驱动"
1. 惠普 LaserJet P1020 Plus 激光打印机概述:
惠普 LaserJet P1020 Plus 是惠普公司针对家庭、个人办公以及小型办公室(SOHO)市场推出的一款激光打印机。这款打印机的设计注重小巧体积和便携操作,适合空间有限的工作环境。其紧凑的设计和高效率的打印性能使其成为小型企业或个人用户的理想选择。
2. 技术特点与性能:
- 预热技术:惠普 LaserJet P1020 Plus 使用了0秒预热技术,能够极大减少打印第一张页面所需的等待时间,首页输出时间不到10秒。
- 打印速度:该打印机的打印速度为每分钟14页,适合处理中等规模的打印任务。
- 月打印负荷:月打印负荷高达5000页,保证了在高打印需求下依然能稳定工作。
- 标配硒鼓:标配的2000页打印硒鼓能够为用户提供较长的使用周期,减少了更换耗材的频率,节约了长期使用成本。
3. 系统兼容性:
驱动程序支持的操作系统包括 Windows Vista 64位版本。用户在使用前需要确保自己的操作系统版本与驱动程序兼容,以保证打印机的正常工作。
4. 市场表现:
惠普 LaserJet P1020 Plus 在上市之初便获得了市场的广泛认可,创下了百万销量的辉煌成绩,这在一定程度上证明了其可靠性和用户对其性能的满意。
5. 驱动程序文件信息:
压缩包内包含了适用于该打印机的官方驱动程序文件 "lj1018_1020_1022-HB-pnp-win64-sc.exe"。该文件是安装打印机驱动的执行程序,用户需要下载并运行该程序来安装驱动。
另一个文件 "jb51.net.txt" 从命名上来看可能是一个文本文件,通常这类文件包含了关于驱动程序的安装说明、版本信息或是版权信息等。由于具体内容未提供,无法确定确切的信息。
6. 使用场景:
由于惠普 LaserJet P1020 Plus 的打印速度和负荷能力,它适合那些需要快速、频繁打印文档的用户,例如行政助理、会计或小型法律事务所。它的紧凑设计也使得这款打印机非常适合在桌面上使用,从而不占用过多的办公空间。
7. 后续支持与维护:
用户在购买后可以通过惠普官方网站获取最新的打印机驱动更新以及技术支持。在安装新驱动之前,建议用户先卸载旧的驱动程序,以避免版本冲突或不必要的错误。
8. 其它注意事项:
- 用户在使用打印机时应注意按照官方提供的维护说明定期进行清洁和保养,以确保打印质量和打印机的使用寿命。
- 如果在打印过程中遇到任何问题,应先检查打印机设置、驱动程序是否正确安装以及是否有足够的打印纸张和墨粉。
综上所述,惠普 LaserJet P1020 Plus 是一款性能可靠、易于使用的激光打印机,特别适合小型企业或个人用户。正确的安装和维护可以确保其稳定和高效的打印能力,满足日常办公需求。
数字电路实验技巧:10大策略,让你的实验效率倍增!
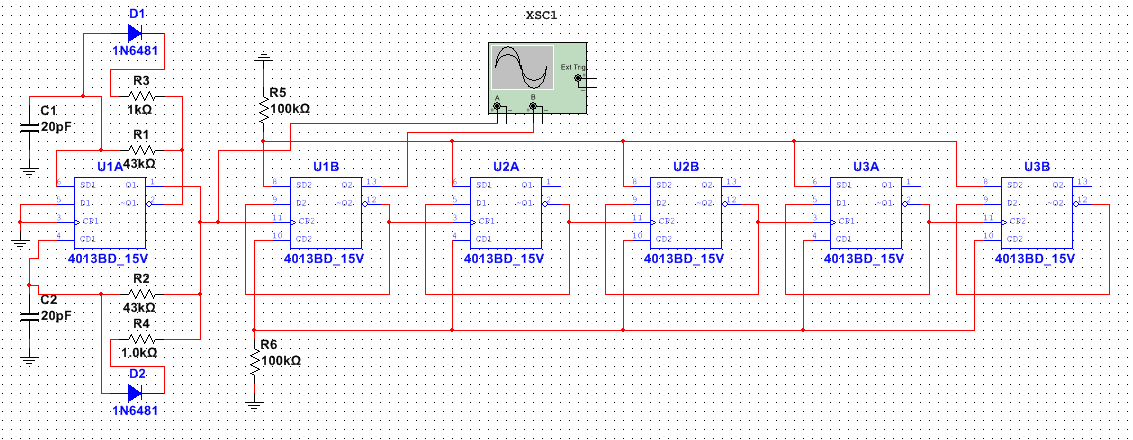
# 摘要
本论文详细介绍了数字电路实验的基础理论、设备使用、设计原则、实践操作、调试与故障排除以及报告撰写与成果展示。首先探讨了数字电路实验所需的基本理论和实验设备的种类与使用技巧,包括测量和故障诊断方法。接着,深入分析了电路设计的原则,涵盖设计流程、逻辑简化、优化策略及实验方案的制定。在实践操作章节中,具体
altium designer布线
### Altium Designer 布线教程和技巧
#### 一、环境设置与准备
为了更高效地完成布线工作,前期的准备工作至关重要。确保原理图已经完全无误并编译成功[^2]。
#### 二、同步查看原理图与PCB布局
通过在原理图标题栏处右键点击并选择 "Split Vertical" 可实现原理图和PCB视图的同时展示,这有助于理解电路连接关系以及提高布线效率。
#### 三、自动布线器配置
Altium Designer内置有强大的自动布线功能。进入“Tools -> PCB Rules and Constraints Editor”,可以自定义诸如最小间距、过孔尺寸等参数来满足
Rust与OpenGL共同打造的迷宫游戏
资源摘要信息:"迷宫游戏开发指南"
在Rust和OpenGL环境下开发迷宫游戏涉及多个方面的知识点,包括编程语言Rust的基本语法和高级特性,OpenGL的图形编程原理以及游戏循环和资源管理等。以下详细说明了这些知识点:
1. Rust编程语言基础
Rust是一种系统编程语言,它提供了内存安全而无需垃圾回收器。Rust的目标是防止空指针解引用、缓冲区溢出等内存安全问题。迷宫游戏开发中,使用Rust可以高效利用系统资源并保证运行时的稳定性和性能。基础知识点包括但不限于:
- 变量和可变性
- 数据类型:整型、浮点型、字符、布尔类型、元组、数组、切片等
- 控制流:if、循环(for, while)、模式匹配
- 函数和闭包
- 所有权、借用和生命周期
- 结构体、枚举和特征
- 模块和使用语句
- 错误处理:Result和Option枚举
- 异步编程:async和await
2. OpenGL图形编程基础
OpenGL(Open Graphics Library)是一个跨语言、跨平台的API,用于渲染2D和3D矢量图形。在Rust中,可以使用gl-rs或其他类似的库来创建OpenGL上下文,并进行渲染操作。迷宫游戏开发中,开发者需要掌握的知识点包括:
- OpenGL上下文的创建和管理
- 着色器语言GLSL的基本语法
- 纹理映射、光源和材质处理
- 几何体的创建和管理(如顶点缓冲、索引缓冲等)
- 渲染管线的各个阶段(顶点处理、裁剪、光栅化等)
- 深度缓冲和模板缓冲的使用
- OpenGL状态机的理解和管理
3. 游戏开发循环
游戏开发循环是指游戏运行时不断循环进行的一系列步骤,通常包括输入处理、游戏状态更新和渲染。迷宫游戏开发中,游戏循环的设计与实现是至关重要的部分。涉及到的知识点包括:
- 游戏状态机的设计
- 输入事件的监听和处理(如键盘、鼠标事件)
- 游戏逻辑的更新(如玩家移动、碰撞检测、迷宫生成逻辑等)
- 场景的渲染和重绘
- 游戏帧率的控制和时间管理
4. 资源管理
资源管理是指游戏中各类资源(如图像、音频、模型等)的加载、使用和释放。在Rust中,这通常涉及到文件读取、内存管理和生命周期控制。迷宫游戏开发中需要的知识点包括:
- 文件系统的操作(如读取迷宫数据文件)
- 内存管理策略(如资源的动态加载和卸载)
- 图像和纹理的加载和使用
- 音频播放控制
- 资源释放时机的确定以避免内存泄漏
5. 迷宫游戏逻辑实现
迷宫游戏的逻辑实现是指游戏中迷宫的生成、玩家的引导和游戏的胜负判定等核心游戏机制。迷宫游戏逻辑实现中的关键知识点包括:
- 迷宫生成算法(如深度优先搜索算法、Prim算法或Kruskal算法等)
- 玩家和游戏对象的移动逻辑
- 路径寻找和导引逻辑(如A*算法)
- 胜负判定和游戏重置逻辑
6. 使用Rust和OpenGL库
实际开发中,开发者会使用一些Rust库来简化OpenGL的调用和管理。相关的知识点包括:
- cargo工具和Rust包管理
- 使用Rust的OpenGL绑定库(如gl-rs、glium等)
- 管理依赖和构建项目的配置文件(Cargo.toml)
- 使用第三方库来处理窗口创建和事件循环(如 glutin)
7. 调试和性能优化
在开发迷宫游戏的过程中,调试和性能优化是重要的环节,以确保游戏运行的流畅性和稳定性。相关的知识点包括:
- 使用调试工具(如gdb、rr、Valgrind等)进行错误追踪和性能分析
- 代码的性能优化策略(如循环展开、内存对齐、缓存优化等)
- 图形渲染的性能优化(如批处理渲染、优化状态切换、减少绘制调用等)
- 使用诊断工具(如Rust的cargo-expand等)来查看代码展开和宏展开
综上所述,Rust和OpenGL迷宫游戏的开发涉及众多知识点,需要开发者具备扎实的编程基础、图形编程经验、游戏开发知识和系统性能优化能力。通过使用Rust的现代编程特性和OpenGL的强大图形处理能力,可以开发出运行高效且稳定的迷宫游戏。
数字电路设计基础:9大技巧带你从理论飞跃到实践
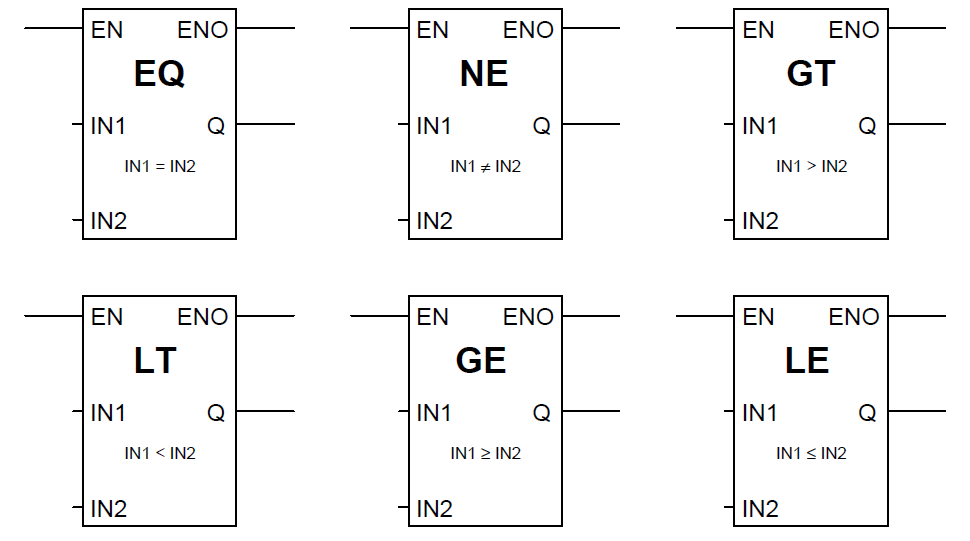
# 摘要
数字电路设计是电子工程领域中的核心部分,它涵盖了从基本概念到高级技巧的广泛知识。本文首先介绍了数字电路设计的基本概念和原理,接着深入探讨了理论基础,包括逻辑门、组合逻辑电路以及时序逻辑电路的设计。随后,文章转向实践应用,讨论了设计工具、仿真测试方法和数字电路在不同领域的应用实例。最后,本
ubuntu 安装opencv2
### Ubuntu 上安装 OpenCV2 版本的库
对于希望在Ubuntu操作系统上安装特定版本如OpenCV2的情况,可以遵循一系列定制化的指令来达成目标。由于当前主流教程多集中于最新版OpenCV及其附加模块(opencv_contrib)的安装指导[^1],针对旧版本(例如OpenCV2)的操作则需特别注意兼容性和依赖关系。
#### 准备工作环境
为了确保顺利安装OpenCV2,在开始前应先更新系统的包列表并升级已有的软件包至最新状态:
```bash
sudo apt update && sudo apt upgrade -y
```
接着,移除任何可能存在的新版本Op