python snv 预处理
时间: 2023-09-18 15:02:00 浏览: 158
Python snv 预处理主要包括以下几个步骤:
1. 数据加载:首先,需要将原始的snv数据读入到Python中进行处理。可以使用Python中的 pandas 库来读取数据文件,该库提供了一种高效的方式来处理和分析大型数据集。
2. 数据清洗:在读取数据之后,需要对数据进行清洗,包括去除无效的数据、处理缺失值和异常值。可以使用 pandas 库中的函数和方法来进行数据清洗操作,如 dropna() 函数去除缺失值、fillna() 函数填充缺失值。
3. 数据转换和特征工程:在清洗数据之后,需要进行数据转换和特征工程,以便为后续的建模和分析做准备。这一步可以包括特征选择、特征标准化、特征编码等操作。可以使用 pandas 中的函数和方法进行特征工程,如 select_dtypes() 函数选择指定数据类型的列、get_dummies() 函数进行独热编码等。
4. 数据集拆分:在进行机器学习建模之前,需要将整个数据集拆分为训练集和测试集。可以使用 sklearn 库中的 train_test_split() 函数来进行数据集的划分。
5. 数据归一化:对于某些机器学习算法,如支持向量机和神经网络,需要对数据进行归一化操作,以确保各个特征之间具有相同的尺度。可以使用 sklearn 中的 MinMaxScaler() 函数来将数据进行归一化。
总结起来,Python snv 预处理包括数据加载、数据清洗、数据转换和特征工程、数据集拆分和数据归一化等步骤。这些步骤的目的是为了准备好规范的数据集,以便于后续的机器学习建模和分析。
相关问题
snv预处理方法python
SNV是Single Nucleotide Variation的缩写,预处理主要目的是将原始数据进行清洗,去除低质量位点和样本,过滤掉常见的单核苷酸多态性(SNP),并进行注释。Python常用的SNV预处理工具包括pysam、vcfpy、pyvcf等。
以下是一个基于pysam的SNV预处理示例代码:
```python
import pysam
# 打开VCF文件
vcf_in = pysam.VariantFile('input.vcf')
# 创建VCF文件
vcf_out = pysam.VariantFile('output.vcf', 'w', header=vcf_in.header)
# 循环遍历每个变异位点
for record in vcf_in:
# 过滤掉质量值小于30的位点和缺失数据过多的样本
if record.qual < 30 or record.num_missing > 10:
continue
# 过滤掉常见的SNP
if record.ref == 'A' and record.alts[0] == 'G':
continue
# 添加注释信息
record.info['DP'] = sum(record.samples[sample]['DP'] for sample in record.samples)
# 写入新的VCF文件
vcf_out.write(record)
# 关闭文件
vcf_in.close()
vcf_out.close()
```
snv光谱预处理python代码
SNV(Standard Normal Variate)光谱预处理是一种常用的光谱预处理方法,可以消除光谱中的散射和背景噪音,提高光谱的信噪比。
在Python中,可以使用scikit-learn库来进行SNV光谱预处理。以下是一个简单的SNV光谱预处理的Python代码示例:
```python
import numpy as np
from sklearn.preprocessing import StandardScaler
def snv_spectra_preprocessing(spectra):
# 计算每个波长的平均值
mean_spectrum = np.mean(spectra, axis=0)
# 对光谱数据进行中心化
centered_spectra = spectra - mean_spectrum
# 计算每个波长的标准差
std_spectrum = np.std(centered_spectra, axis=0)
# 计算每个样本的标准差
std_samples = np.std(centered_spectra, axis=1)
# 对光谱数据进行缩放,除以每个波长的标准差,乘以每个样本的标准差
snv_spectra = centered_spectra / std_spectrum * std_samples[:, np.newaxis]
return snv_spectra
# 使用示例
spectra = np.array([[1, 2, 3], [4, 5, 6], [7, 8, 9]]) # 示例光谱数据,假设包含3个样本,每个样本有3个波长
snv_spectra = snv_spectra_preprocessing(spectra)
print(snv_spectra)
```
在上述代码中,`snv_spectra_preprocessing`函数接受一个包含光谱数据的二维数组作为输入。该函数首先计算每个波长的平均值和标准差,然后对光谱数据进行中心化操作,再对数据进行缩放操作以实现SNV预处理。最后,返回经过SNV预处理后的光谱数据。
在示例中,我们定义了一个3x3的光谱数据矩阵,然后调用`snv_spectra_preprocessing`函数进行SNV光谱预处理,并打印预处理后的光谱数据。
请注意,上述代码中只是对光谱数据进行了简单的SNV处理,实际应用中可能需要更复杂的处理步骤,例如使用不同的波长范围、对数转换等。根据具体问题和数据,可以对代码进行调整和扩展。
阅读全文
相关推荐
大家在看
MOOC工程伦理课后习题答案(主观+判断+选择)期末考试答案.docx
MOOC工程伦理课程,课程讲义以及课后选择题、判断题和主观题习题答案
基于Farrow结构的滤波器频响特性matlab仿真,含仿真操作录像
1.版本:matlab2022a,包含仿真操作录像,操作录像使用windows media player播放。
2.领域:Farrow滤波器。
3.内容:基于Farrow结构的滤波器频响特性matlab仿真
% 得到Farrow结构滤波器的频响特性
for j=1:Nfil
x=(j-1)*xinc + 0.0001; % 避免出现sin(0)/0
h = C(Np+1,:); % 由拟合后的子滤波器系数矩阵
for n=1:Np
h=h+x^n*C(Np+1-n,:); % 得到子滤波器的系数和矩阵
end
h=h/sum(h); % 综合滤波器组的系数矩阵
H = freqz(h,1,wpi);
mag(j,:) = abs(H);
end
plot(w,20*log10(abs(H)));
grid on;xlabel('归一化频率');ylabel('幅度');
4.注意事项:注意MATLAB左侧当前文件夹路径,必须是程序所在文件夹位置,具体可以参考视频录。
电路ESD防护原理与设计实例.pdf
电路ESD防护原理与设计实例,不错的资源,硬件设计参考,相互学习
主生產排程員-SAP主生产排程
主生產排程員
比較實際需求與預測需求,提出預測與MPS的修訂建議。
把預測與訂單資料轉成MPS。
使MPS能配合出貨與庫存預算、行銷計畫、與管理政策。
追蹤MPS階層產品安全庫存的使用、分析MPS項目生產數量和FAS消耗數量之間的差異、將所有的改變資料輸入MPS檔案,以維護MPS。
參加MPS會議、安排議程、事先預想問題、備好可能的解決方案、將可能的衝突搬上檯面。
評估MPS修訂方案。
提供並監控對客戶的交貨承諾。
信息几何-Information Geometry
信息几何是最近几年新的一个研究方向,主要应用于统计分析、控制理论、神经网络、量子力学、信息论等领域。本书为英文版,最为经典。阅读需要一定的英文能力。
最新推荐
开发板基于STM32H750VBT6+12位精度AD9226信号采集快速傅里叶(FFT)变计算对应信号质量,资料包含原理图、调试好的源代码、PCB文件可选
开发板基于STM32H750VBT6+12位精度AD9226信号采集快速傅里叶(FFT)变计算对应信号质量,资料包含原理图、调试好的源代码、PCB文件可选
基于plc的加工站传送包装站控制系统设计加工传送包装站电气控制 带解释的梯形图程序,接线图原理图图纸,io分配,组态画面 红旗hot界面多种组态可供选择,详情请点头像查看
基于plc的加工站传送包装站控制系统设计加工传送包装站电气控制
带解释的梯形图程序,接线图原理图图纸,io分配,组态画面
[红旗][hot]界面多种组态可供选择,详情请点头像查看
H.264高分辨率视频会议中的自适应比特率控制算法研究与应用
内容概要:本文提出了一种名为动态常量速率因子(DCRF)的新颖率控算法,用于解决当前基于x264编码器的标准H.264高分辨率(HD)视频会议系统无法适应非专用网络的问题。该算法能够动态调整视频流的比特率,以匹配不同网络带宽情况下的传输需求,从而提供高质量的实时视频传输体验。文章还探讨了传统平均比特率(ABR)以及恒定速率因子(CRF)两种常用算法的优缺点,在此基础上改进得出了更适配于实时性的新方法DCRF,它能迅速对网络状态变化做出响应并稳定视频质量。为了验证这一方法的有效性和优越性,实验采用了主观测试与客观指标相结合的方式进行了全面评估。实测数据表明,新的率控制器可以在有限的带宽下提供更佳的用户体验。
适用人群:视频编解码、视频会议系统、多媒体通信领域的研究人员和技术专家;对于高带宽视频传输解决方案感兴趣的专业人士;希望深入了解视频压缩标准及其性能特点的人士。
使用场景及目标:适用于所有需要进行高清视频通话或多方视频协作的情境;主要应用于互联网环境下,特别是存在不确定因素影响实际可用带宽的情况下;目标是确保即使在网络不稳定时也能维持较好的画质表现,减少卡顿、延迟等问题发生。
其他说明:论文不仅提供了理论分析和技术细节,还包括具体的参数配置指导和大量的实验数据分析。这有助于开发者将此算法融入现有的视频处理框架之中,提高系统的鲁棒性和效率。同时,研究中所涉及的一些概念如率失真优化、组间预测误差模型等也值得深入探究。
西门子S7一1200 PLc程序项目,cPU1214和ET200 iO站点,博途V16与V17版,HMi为kTP1200.模拟量转,电动阀控制,液位控制,Modbus通讯控制变频器,Pid控制,PU
西门子S7一1200 PLc程序项目,cPU1214和ET200 iO站点,博途V16与V17版,HMi为kTP1200.模拟量转,电动阀控制,液位控制,Modbus通讯控制变频器,Pid控制,PUt与get指令,汅水处理项目
三菱PLC转盘机程序 此程序已经实际设备上生产应用,程序成熟可靠,借鉴价值高,程序有注释,用的三菱FX5Uplc,带6根轴,视觉判定ok还是NG 是入门级三菱PLC电气爱好从业人员借鉴和参考经
三菱PLC转盘机程序
此程序已经实际设备上生产应用,程序成熟可靠,借鉴价值高,程序有注释,用的三菱FX5Uplc,带6根轴,视觉判定ok还是NG。
是入门级三菱PLC电气爱好从业人员借鉴和参考经典案列。
程序.触摸屏·IO表
海康无插件摄像头WEB开发包(20200616-20201102163221)
资源摘要信息:"海康无插件开发包"
知识点一:海康品牌简介
海康威视是全球知名的安防监控设备生产与服务提供商,总部位于中国杭州,其产品广泛应用于公共安全、智能交通、智能家居等多个领域。海康的产品以先进的技术、稳定可靠的性能和良好的用户体验著称,在全球监控设备市场占有重要地位。
知识点二:无插件技术
无插件技术指的是在用户访问网页时,无需额外安装或运行浏览器插件即可实现网页内的功能,如播放视频、音频、动画等。这种方式可以提升用户体验,减少安装插件的繁琐过程,同时由于避免了插件可能存在的安全漏洞,也提高了系统的安全性。无插件技术通常依赖HTML5、JavaScript、WebGL等现代网页技术实现。
知识点三:网络视频监控
网络视频监控是指通过IP网络将监控摄像机连接起来,实现实时远程监控的技术。与传统的模拟监控相比,网络视频监控具备传输距离远、布线简单、可远程监控和智能分析等特点。无插件网络视频监控开发包允许开发者在不依赖浏览器插件的情况下,集成视频监控功能到网页中,方便了用户查看和管理。
知识点四:摄像头技术
摄像头是将光学图像转换成电子信号的装置,广泛应用于图像采集、视频通讯、安全监控等领域。现代摄像头技术包括CCD和CMOS传感器技术,以及图像处理、编码压缩等技术。海康作为行业内的领军企业,其摄像头产品线覆盖了从高清到4K甚至更高分辨率的摄像机,同时在图像处理、智能分析等技术上不断创新。
知识点五:WEB开发包的应用
WEB开发包通常包含了实现特定功能所需的脚本、接口文档、API以及示例代码等资源。开发者可以利用这些资源快速地将特定功能集成到自己的网页应用中。对于“海康web无插件开发包.zip”,它可能包含了实现海康摄像头无插件网络视频监控功能的前端代码和API接口等,让开发者能够在不安装任何插件的情况下实现视频流的展示、控制和其他相关功能。
知识点六:技术兼容性与标准化
无插件技术的实现通常需要遵循一定的技术标准和协议,比如支持主流的Web标准和兼容多种浏览器。此外,无插件技术也需要考虑到不同操作系统和浏览器间的兼容性问题,以确保功能的正常使用和用户体验的一致性。
知识点七:安全性能
无插件技术相较于传统插件技术在安全性上具有明显优势。由于减少了外部插件的使用,因此降低了潜在的攻击面和漏洞风险。在涉及监控等安全敏感的领域中,这种技术尤其受到青睐。
知识点八:开发包的更新与维护
从文件名“WEB无插件开发包_20200616_20201102163221”可以推断,该开发包具有版本信息和时间戳,表明它是一个经过时间更新和维护的工具包。在使用此类工具包时,开发者需要关注官方发布的版本更新信息和补丁,及时升级以获得最新的功能和安全修正。
综上所述,海康提供的无插件开发包是针对其摄像头产品的网络视频监控解决方案,这一方案通过现代的无插件网络技术,为开发者提供了方便、安全且标准化的集成方式,以实现便捷的网络视频监控功能。
PCNM空间分析新手必读:R语言实现从入门到精通

# 摘要
本文旨在介绍PCNM空间分析方法及其在R语言中的实践应用。首先,文章通过介绍PCNM的理论基础和分析步骤,提供了对空间自相关性和PCNM数学原理的深入理解。随后,详细阐述了R语言在空间数据分析中的基础知识和准备工作,以及如何在R语言环境下进行PCNM分析和结果解
生成一个自动打怪的脚本
创建一个自动打怪的游戏脚本通常是针对游戏客户端或特定类型的自动化工具如Roblox Studio、Unity等的定制操作。这类脚本通常是利用游戏内部的逻辑漏洞或API来控制角色的动作,模拟玩家的行为,如移动、攻击怪物。然而,这种行为需要对游戏机制有深入理解,而且很多游戏会有反作弊机制,自动打怪可能会被视为作弊而被封禁。
以下是一个非常基础的Python脚本例子,假设我们是在使用类似PyAutoGUI库模拟键盘输入来控制游戏角色:
```python
import pyautogui
# 角色位置和怪物位置
player_pos = (0, 0) # 这里是你的角色当前位置
monster
CarMarker-Animation: 地图标记动画及转向库
资源摘要信息:"CarMarker-Animation是一个开源库,旨在帮助开发者在谷歌地图上实现平滑的标记动画效果。通过该库,开发者可以实现标记沿路线移动,并在移动过程中根据道路曲线实现平滑转弯。这不仅提升了用户体验,也增强了地图应用的交互性。
在详细的技术实现上,CarMarker-Animation库可能会涉及到以下几个方面的知识点:
1. 地图API集成:该库可能基于谷歌地图的API进行开发,因此开发者需要有谷歌地图API的使用经验,并了解如何在项目中集成谷歌地图。
2. 动画效果实现:为了实现平滑的动画效果,开发者需要掌握CSS动画或者JavaScript动画的实现方法,包括关键帧动画、过渡动画等。
3. 地图路径计算:标记在地图上的移动需要基于实际的道路网络,因此开发者可能需要使用路径规划算法,如Dijkstra算法或者A*搜索算法,来计算出最合适的路线。
4. 路径平滑处理:仅仅计算出路线是不够的,还需要对路径进行平滑处理,以使标记在转弯时更加自然。这可能涉及到曲线拟合算法,如贝塞尔曲线拟合。
5. 地图交互设计:为了与用户的交互更为友好,开发者需要了解用户界面和用户体验设计原则,并将这些原则应用到动画效果的开发中。
6. 性能优化:在实现复杂的动画效果时,需要考虑程序的性能。开发者需要知道如何优化动画性能,减少卡顿,确保流畅的用户体验。
7. 开源协议遵守:由于CarMarker-Animation是一个开源库,开发者在使用该库时,需要遵守其开源协议,合理使用代码并遵守贡献指南。
此库的文件名'CarMarker-Animation-master'表明这是一个主分支的项目,可能包含源代码文件、示例项目、文档说明等资源。开发者可以通过下载解压缩后获得这些资源,并根据提供的文档来了解如何安装和使用该库。在使用过程中,建议仔细阅读开源项目的贡献指南和使用说明,以确保库的正确集成和使用,同时也可以参与开源社区,与其他开发者共同维护和改进这一项目。"
5G核心网元性能瓶颈揭秘
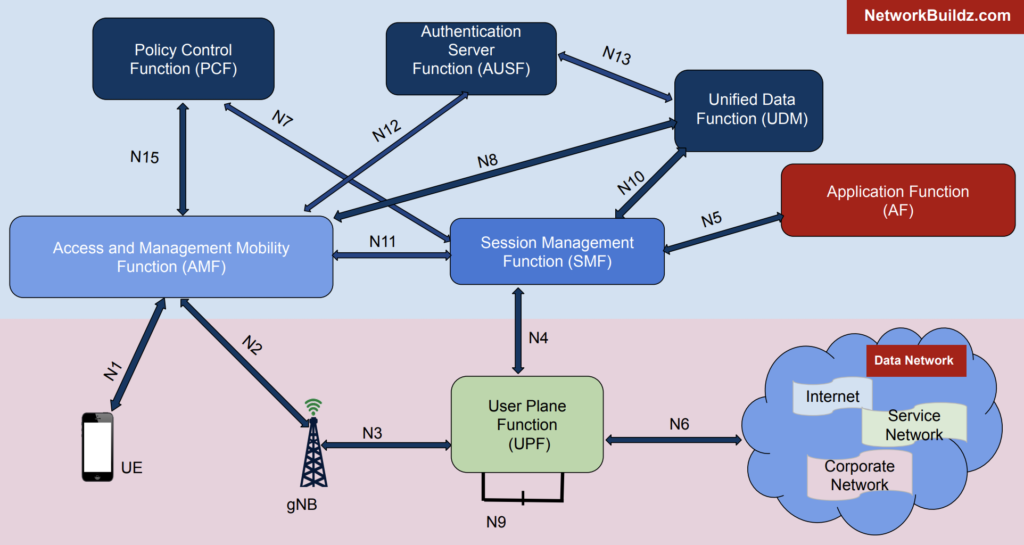
# 摘要
随着5G技术的发展和应用,其核心网的性能优化成为了行业关注的焦点。本文首先概述了5G核心网的架构,并对性能瓶颈进行深入分析,识别了关键的性能指标和瓶颈识别方法。通过案例分析,展示了核心网元常见的性能问题及其诊断和解决过程。随后,文章提出了多项性能优化策略,包括网络设计、系统配置调整以及新技术的应用。此外,本文探讨了安全挑战如何影响核心网的性能,