优化光储材料:石墨烯-偶氮苯衍生物的分子模型与DFT计算
版权申诉
176 浏览量
更新于2024-07-02
收藏 1.9MB PDF 举报
云计算与光储热材料的研究在当前科技领域日益受到关注,特别是利用光致变色分子如偶氮苯(azobenzene)来开发高效的太阳能热能存储材料。传统的孤立偶氮苯分子因其独特的可逆顺/反异构化性质被看作是这一领域的潜在关键成分。然而,传统偶氮苯的能量存储能力较低,其转化能垒仅为0.58电子伏特(eV),且热稳定性能不理想,无法满足高性能太阳能热材料的需求。
为了提高偶氮苯的性能,研究者们设计了一系列偶氮苯衍生物,并将其通过共价键结合到碳材料表面,如碳纳米管或石墨烯上。这种策略旨在通过形成高度有序的结构,调控偶氮苯分子间的相互作用力以及立体位阻,从而控制顺式和反式异构体之间的能量差异。目标是实现偶氮苯-碳纳米复合材料的高能量存储特性。
本文特别聚焦于将不同取代基(如羧酸基、羟基、甲氧基、硝基等)的偶氮苯作为光学响应单元,选择性地锚定在二维石墨烯表面,构建太阳能热能分子模型。作者采用第一性原理方法,基于密度泛函理论(DFT)进行计算,详细分析了这些分子的能级,包括顺式和反式异构体的理论能量差,以期优化偶氮苯在光照下的反应动力学和热稳定性。
通过这种方式,研究人员能够深入理解偶氮苯分子在石墨烯表面的行为,揭示其与环境交互时的能量转移和储存机制。这不仅有助于提升太阳能热能材料的性能,也为未来设计新型光化学转换和存储系统提供了宝贵的理论指导。随着计算模拟技术的发展,这种理论计算在指导实验设计和优化实际应用中的作用将越来越大。
第一章 绪论
5
1.4.2 热能的存储
(1)相变材料储能
[32]
(PCMs) 相变过程一般是等温或近似等温过程,随
温度变化而改变形态并能提供潜热的物质。PCMs 由固态变为液态或由液态变为
固态的过程称为相变过程,这时相变材料将吸收或释放大量的潜热。PCMs 按材
料的组成可以分为三类:有机类
[33]
(链烷烃、脂肪酸等,相变潜热在 10~300
kJ/kg)、无机类(水合物、熔融盐、金属等,20~250 kJ/kg)和有机-无机
[34]
(有
机-有机、有机-无机、无机-无机,100~200 kJ/kg)复合相变储能材料。由此可以
看出来复合相变储能材料能降低熔点,并且温度变化维持的范围小,相变潜热密
度高在经济效益上具有较高的竞争优势等特点。
(2)吸附储能 吸附是通过一种固体或液体的作为吸附剂俘获气体或蒸汽的
过程
[35]
。吸附过程包括范德华力的热物理反应
[36]
(物理吸附)和共价力的热化
学反应(化学吸附)。化学吸附过程单位质量下的储热比物理吸附大得多,但是
不可逆。当对系统加热时,吸着物/吸附剂分解为两种物质以化学势的形式分别
存储起来,并能长期存储;当两种物质混合时,能量释放。吸附材料在重复存储
介质中具有相当高的存储密度,对于太阳能的存储具有非常大的实用价值。
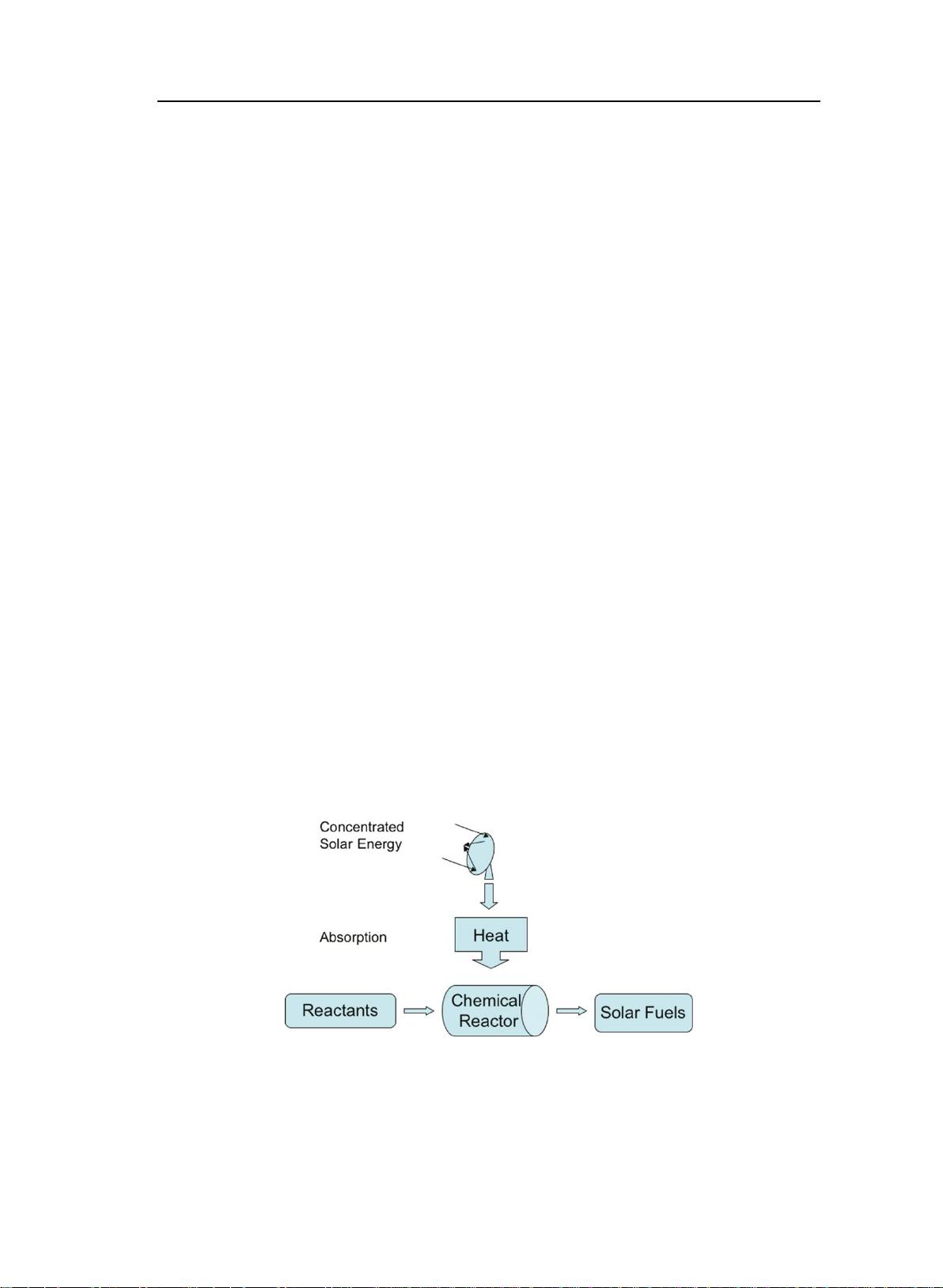
(3)太阳能燃料 太阳能燃料的工作原理如图 1-1 所示,用光学设备将太阳
能集中起来,用吸热材料将其转变为储存起来的可转移的太阳能燃料。太阳能燃
料最具有竞争力的优势在于理论上太阳能的存储和释放的循环过程中没有能量
的损耗,能达到 100%的利用率。许多材料可用于太阳能燃料材料,例如太阳能
氢燃料
[37]
、太阳能金属
[38]
、太阳能化学热管
[39]
和太阳能光热燃料
[40]
等。
图 1-1 太阳能燃料生产过程示意图
Figure 1-1 Diagram of solar fuels generation
1)太阳能氢燃料 用于燃烧氢气产生能量的仪器叫氢引擎。利用太阳能产生
氢气的方法有三种:热化学法制氢、光电化学分解法制氢、光催化法制氢、人工
万方数据


剩余56页未读,继续阅读
1104 浏览量
129 浏览量
点击了解资源详情
2021-09-25 上传
2021-09-15 上传
2021-09-25 上传
2021-09-25 上传
148 浏览量

programxh
- 粉丝: 17
- 资源: 1万+
 我的内容管理
展开
我的内容管理
展开







最新资源
- xxl-job.rar
- org-transclusion:(alpha)Emacs软件包,用于通过组织模式启用转写
- 基于ASP.net高校网上教材征订系统的设计与实现(源代码+论文).rar
- 数据分析统计图表ppt模板
- 基于MATLAB实现的BP神经网络的非线性系统建模非线性函数拟合(Maltab源代码+数据集+运行说明).zip
- RAD Studio 10.4.1 KeyPatch
- NScache-开源
- android-ndk-r19c-windows-x86_64.zip
- ember-swagger-ui:Ember插件,可快速轻松地将swagger-ui添加到您的Ember App
- 宝米勒 MC200T系列变频器用户手册v2.0.zip
- iOS美白/灰色/旋转/合成图片(添加文字)
- 易语言源码Access数据库中的数据导出到Excel中.rar
- koa-preprocessor
- ember-cli-updater:ember-cli插件,可帮助您更新ember-cli应用程序或插件
- Practice
- 暂时的
