"宏基因组多样性分析与可视化培训及环境配置技巧"
需积分: 0 103 浏览量
更新于2023-12-16
收藏 5.5MB PDF 举报
本文主要介绍了宏基因组(meta-genome)的多样性分析和可视化方法,并提供了相关的易生信宏基因组R语言包的安装和常见问题解答。本文内容来源于2021年5月27日中国肠道大会上刘永鑫中科院遗传发育所的演讲和宏基因组公众号的创始人。同时,还介绍了一些在Windows系统下使用R和RStudio的设置方法。
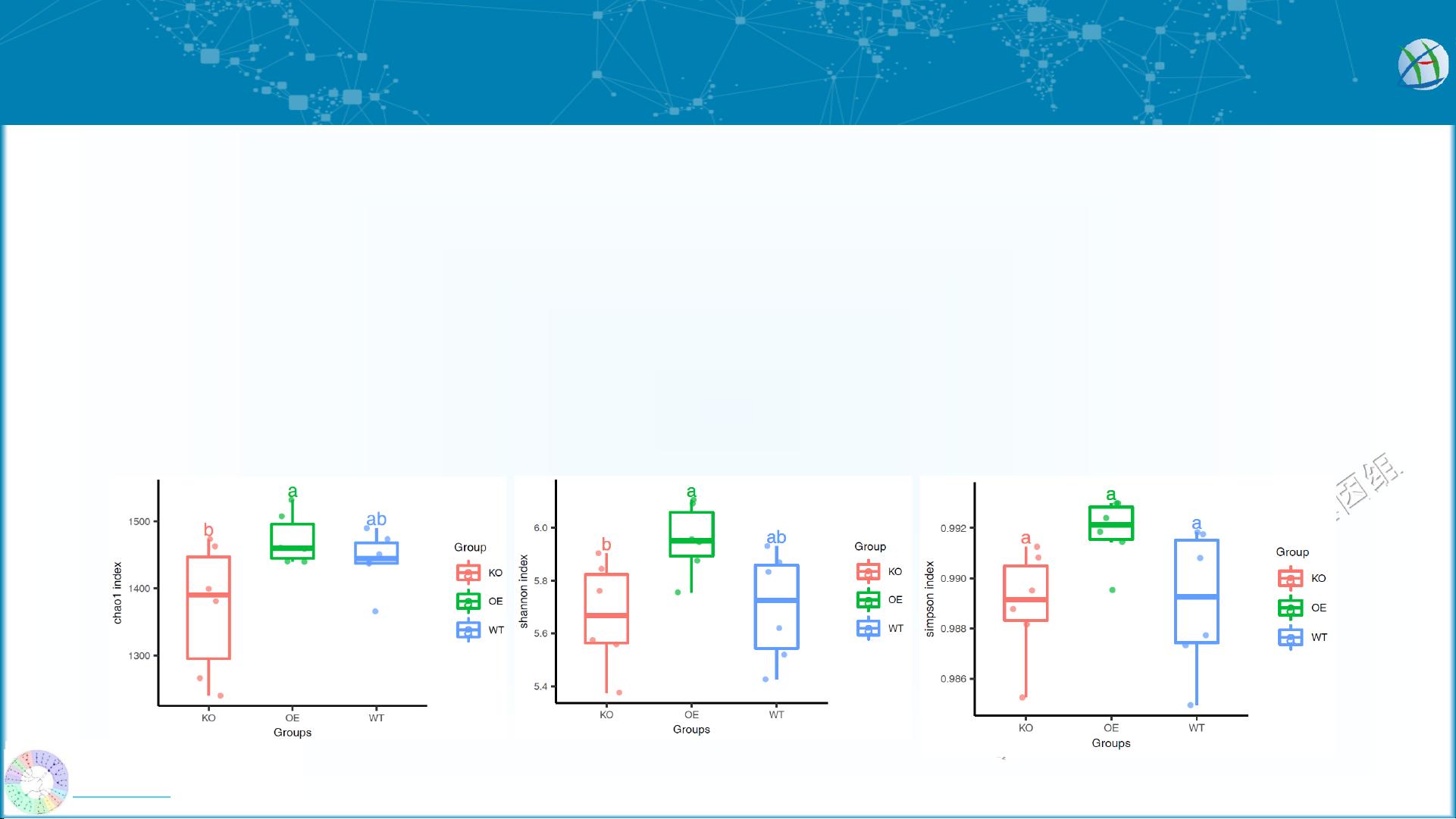
宏基因组是对环境中微生物群体的基因组DNA进行测序和分析的一种方法,该方法能够揭示微生物群体的组成、功能和互作关系等信息。多样性分析和可视化是宏基因组研究中常用的方法,可以帮助研究人员更好地理解和解释微生物群体的多样性特征。本文提供的方法和技巧可以帮助读者在宏基因组研究中进行多样性分析和可视化。
在多样性分析方面,本文介绍了易生信宏基因组R语言包的使用方法。首先,读者需要在Windows系统中安装R语言的环境。具体方法是将提供的4.0.zip压缩包解压至文档中R\win-library目录。然后,在RStudio中进行几个设置,如解决中文乱码问题、设置默认文本编码为UTF-8以及选择CRAN镜像等。这些设置可以帮助读者更好地使用R语言进行宏基因组数据分析。
在可视化方面,本文介绍了宏基因组数据的图形预览方法。通过使用易生信宏基因组R语言包,读者可以将宏基因组数据转化为图形,并进行可视化展示。宏基因组数据的可视化可以帮助研究人员直观地观察微生物群体的组成和分布情况,进一步了解宏基因组的多样性特征。
此外,本文还解答了一些常见的R语言包安装和使用问题。例如,如何批量安装R语言包、如何加速安装包下载以及如何处理命令行类型等。这些问题的解答可以帮助读者更好地解决在宏基因组研究中常遇到的技术困难,提高数据分析的效率和准确性。
总而言之,本文介绍了宏基因组(meta-genome)的多样性分析和可视化方法,并提供了宏基因组R语言包的安装和常见问题解答。这些方法和技巧可以帮助读者更好地进行宏基因组研究,深入了解微生物群体的多样性特征。同时,本文还提供了一些在Windows系统下使用R和RStudio的设置方法,帮助读者更好地使用R语言进行宏基因组数据分析。总之,本文为宏基因组研究人员提供了一些有用的工具和资源,希望对他们的研究工作有所帮助。


2022-08-03 上传
2024-06-20 上传
点击了解资源详情
点击了解资源详情
点击了解资源详情
2021-05-12 上传
2024-05-27 上传
2024-03-02 上传

大禹倒杯茶
- 粉丝: 23
- 资源: 331
 我的内容管理
展开
我的内容管理
展开







最新资源
- Java集合ArrayList实现字符串管理及效果展示
- 实现2D3D相机拾取射线的关键技术
- LiveLy-公寓管理门户:创新体验与技术实现
- 易语言打造的快捷禁止程序运行小工具
- Microgateway核心:实现配置和插件的主端口转发
- 掌握Java基本操作:增删查改入门代码详解
- Apache Tomcat 7.0.109 Windows版下载指南
- Qt实现文件系统浏览器界面设计与功能开发
- ReactJS新手实验:搭建与运行教程
- 探索生成艺术:几个月创意Processing实验
- Django框架下Cisco IOx平台实战开发案例源码解析
- 在Linux环境下配置Java版VTK开发环境
- 29街网上城市公司网站系统v1.0:企业建站全面解决方案
- WordPress CMB2插件的Suggest字段类型使用教程
- TCP协议实现的Java桌面聊天客户端应用
- ANR-WatchDog: 检测Android应用无响应并报告异常
