使用MapReduce框架加速大规模生物数据集计算
64 浏览量
更新于2024-08-29
收藏 1.64MB PDF 举报
"加速大型生物数据集计算利用MapReduce框架"
在生物信息学领域,随着高通量测序技术的发展,产生了海量的生物学数据。这些数据包括基因组序列、转录组表达谱、蛋白质相互作用网络等,它们为揭示生命现象提供了前所未有的深度和广度。然而,这些大数据的分析与挖掘工作面临着巨大的计算挑战。本文“使用最大信息系数(Maximal Information Coefficient, MIC)在生物信息学中分析大型生物数据集”探讨了一种利用MapReduce框架来加速计算的方法,尤其针对基因组序列和生物注释中的 MIC 计算。
最大信息系数(MIC)是一种衡量两个变量之间关系强度和非线性关联性的统计指标。它在生物信息学中有着广泛的应用,例如在寻找基因与疾病之间的关联、表观遗传标记与基因表达模式的关系等。然而,由于生物数据的规模巨大,传统的串行计算方法计算 MIC 的效率极低,严重影响了研究进度。
MapReduce 是一种分布式计算模型,由Google提出,旨在处理和生成大规模数据集。该模型将复杂的计算任务分解成两个主要阶段:Map阶段和Reduce阶段。Map阶段将输入数据分割成小块,然后在多台计算机上并行处理;Reduce阶段则对Map阶段的结果进行整合,输出最终结果。这种并行处理能力使得MapReduce非常适合处理大规模生物数据集的计算任务。
在这篇研究论文中,作者Chao Wang等人设计了一种基于MapReduce的并行算法,用于加速MIC的计算过程。他们首先将数据集划分为多个子集,然后在Map阶段计算每个子集内的MIC值,最后在Reduce阶段聚合所有子集的结果,得到全局的MIC值。这种方法充分利用了分布式计算的优势,显著提高了计算效率,同时保持了结果的准确性。
论文还可能涉及了如何有效地划分数据、如何优化Map和Reduce阶段的计算逻辑,以及如何处理数据的缺失值和异常值等问题。通过实验,作者验证了他们的方法在处理大型生物数据集时,相比于传统方法有显著的性能提升,并且在资源消耗上更为优化。
此外,论文可能会讨论这种并行计算方法在实际生物信息学应用中的适用性,比如在全基因组关联研究(GWAS)、单细胞测序数据分析等方面的效果,以及如何将这种方法与其他生物信息学工具和软件结合,构建更高效的数据分析流程。
这篇论文为生物信息学领域的研究人员提供了一个实用的解决方案,有助于他们快速有效地挖掘大规模生物学数据中的潜在关联,推动了生物医学研究的进展。这一工作强调了并行计算技术在应对生物信息学挑战中的重要作用,并为未来相关领域的研究奠定了基础。
1545-5963 (c) 2015 IEEE. Personal use is permitted, but republication/redistribution requires IEEE permission. See http://www.ieee.org/publications_standards/publications/rights/index.html for more information.
This article has been accepted for publication in a future issue of this journal, but has not been fully edited. Content may change prior to final publication. Citation information: DOI 10.1109/TCBB.2016.2550430, IEEE/ACM
Transactions on Computational Biology and Bioinformatics
WANG ET AL.: ANALYZING LARGE BIOLOGICAL DATASETS IN BIOINFORMATICS WITH MAXIMAL INFORMATION COEFFICIENT 3
[15-18]. Of these literatures, how to identify the data cor-
relation has posed significant challenges to the academic
and industrial researchers. There have been a wide range
of methods for identifying interesting relationships be-
tween pairs of variables in large data sets in bioinformat-
ics [19], including methods formulated around the axio-
matic framework for measures of dependence [20], other
state-of-the-art measures of dependence, and several
nonparametric estimation techniques that can be used to
score pairs of variables based on the relationship of the
estimated curve. Methods such as splines and regression
estimators [21] tend to be equitable across functional rela-
tionships but they fail to find many simple and important
types of relationships that are not functional. Although
these methods are not intended to provide generality,
most of them are unsuitable for identifying all potentially
interesting relationships in a large scale data set. Similar
methods such as mutual information estimators, maximal
correlation, principal curve, distance correlation, and the
spearman rank correlation coefficient methods are able to
detect broader classes of relationships. However, these
methods are not equitable even in the basic case of func-
tional relationships: They show a strong preference for
some types of functions, even at identical noise levels. For
example, Reshef [6] has established the generality of max-
imal information coefficient (MIC) through proofs, show-
ing its equitability on functional relationships through
simulations, and observe the intuitively equitable behav-
ior on more general associations.
Although researchers have proven MIC is a creditable
approach to detect the relationships between variable
pairs, it still consumes significant time for analyzing large
scale data set due to the complex calculation process.
With respect to the optimization approaches, GPU and
FPGA based approaches are two dominant methodolo-
gies in heterogeneous architecture design paradigms. For
example, RapidMic [22] is a cross-platform tool for the
rapid computation of the maximal information coefficient
based on parallel computing methods. Through parallel
processing, it can effectively analyze the large-scale bio-
logical datasets with a remarkable reduced computing
time. Similarly a simulated annealing and genetic algo-
rithm was developed [23] to facilitate the optimal calcula-
tion of MIC, and the convergence of SG was proved based
on Markov theory. Lopez-Paz et.al [24] introduce the ran-
domized dependence coefficient, which is a measure of
nonlinear dependence between random variables of arbi-
trary dimension. Kinney et. al [25] identify artifacts in the
reported simulation, in particular for the estimates of mu-
tual information when these artifacts are removed. Re-
cently Nature Biotechnology [26] solicits comments from
several practitioners versed in data-intensive biological
research. Their responses not only highlight the appeal of
methods like MIC for biological research, but also raise
some important reservations as to its widespread use and
statistical power. Paninski [27] presents some results on
the nonparametric estimation of entropy and mutual in-
formation. Kraskov et. al [28] present two classes of im-
proved estimators for mutual information, from samples
of random points distributed according to some joint
probability density. To show the effectiveness of MIC in
medical imaging field, Pluim et.al [29] summarize the
MIC based registration of medical images. Reshef et.al [30]
present the MIC calculation with more comprehensive
understanding to show the effectiveness and efficiency.
Similarly, many scientific applications have been opti-
mized by GPU and FPGA accelerators, such as [31], [5]
and [16]. In particular, these approaches consist of a series
of nodes, each of which has both a CPU controller and a
heterogeneous accelerator. All nodes are under the con-
trolling of the scheduler that is responsible for issuing
tasks and balancing workloads, which increase the design
complexity and burden of the software programmers.
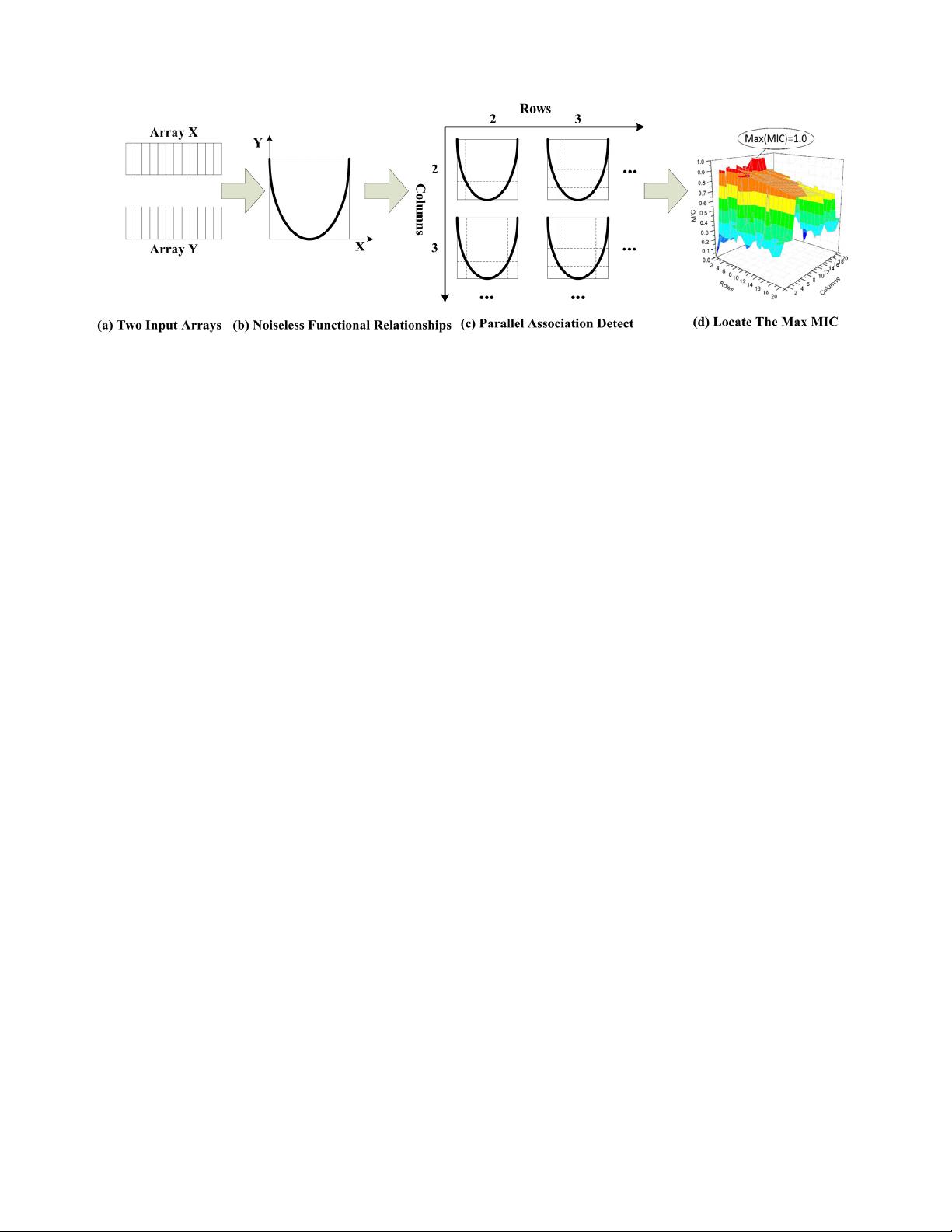
3 MIC CALCULATION ALGORITHM
To reduce the computation complexity in the original
algorithm, Reshef [6] has introduced a dynamic pro-
gramming improvement.
In this section, we will
demonstrate the algorithm first, and then analyze the
strategy of parallelization using MapReduce frame-
work on multiple computing machines
.
3.1. Original Algorithm Description
In particular, the MINE algorithm is designed for heu-
ristically generating the characteristic matrix of two-
variable data sets. To calculate MIC matrix, the algo-
rithm would ideally optimize over all possible grids.
Figure 2. Detailed process of MINE algorithm description
剩余13页未读,继续阅读
2021-02-21 上传
2022-01-23 上传
2021-02-11 上传
2023-09-01 上传
2022-03-07 上传
2021-02-11 上传
2021-02-21 上传
2016-02-26 上传
点击了解资源详情
点击了解资源详情

weixin_38595473
- 粉丝: 3
- 资源: 875
 我的内容管理
展开
我的内容管理
展开







最新资源
- BottleJS快速入门:演示JavaScript依赖注入优势
- vConsole插件使用教程:输出与复制日志文件
- Node.js v12.7.0版本发布 - 适合高性能Web服务器与网络应用
- Android中实现图片的双指和双击缩放功能
- Anum Pinki英语至乌尔都语开源词典:23000词汇会话
- 三菱电机SLIMDIP智能功率模块在变频洗衣机的应用分析
- 用JavaScript实现的剪刀石头布游戏指南
- Node.js v12.22.1版发布 - 跨平台JavaScript环境新选择
- Infix修复发布:探索新的中缀处理方式
- 罕见疾病酶替代疗法药物非临床研究指导原则报告
- Node.js v10.20.0 版本发布,性能卓越的服务器端JavaScript
- hap-java-client:Java实现的HAP客户端库解析
- Shreyas Satish的GitHub博客自动化静态站点技术解析
- vtomole个人博客网站建设与维护经验分享
- MEAN.JS全栈解决方案:打造MongoDB、Express、AngularJS和Node.js应用
- 东南大学网络空间安全学院复试代码解析
