机器学习驱动的团簇能量预测与结构优化研究
下载需积分: 0 | PDF格式 | 1.86MB |
更新于2024-06-30
| 183 浏览量 | 举报
这篇论文主要探讨了利用机器学习技术来解决团簇能量预测和结构全局寻优的问题,重点关注了金属团簇(如金团簇Au)和硼团簇B的优化。团簇是材料科学中的重要研究对象,找到它们的全局最优结构有助于新材料的设计和性能探索。传统的理论计算方法在效率和精度上存在不足,而机器学习可以提供更高效的学习和计算手段。
针对问题1,论文中提到传统金属团簇能量模型的局限性,并构建了一个包含变异因子的Sutton Chen能量预测模型。通过改进粒子群优化算法(PSO),引入自适应惯性权重,提高了搜索效率,避免了陷入局部最优。实验结果显示,对于20个金原子组成的团簇(Au20),最低能量值为-1557.8251,对应的最优结构为对称性的正四面体。
问题2聚焦于32个金原子的团簇(Au32)。论文提出了多条件约束的异构体生成方法,结合AWPSO算法,进行结构的三维重构。经过预测和优化,找到了一个类笼型结构,其最低能量值为-2544.3431,显示出较高的稳定性。
在问题3中,由于传统模型难以准确描述硼团簇(B)的能量,研究团队采用了基于库伦矩阵本征谱的机器学习模型。他们结合AWPSO算法优化了适应度函数,提出了一种新的全局寻优方法。结果显示,45个硼原子组成的团簇(B45)的最低能量结构为带一定曲率的类平面结构,中心有一个六边形空位,能量值为-114305.0794。
最后,在问题4中,研究人员改进了之前的寻优策略,引入库伦矩阵本征谱并开发了多条件约束的NN-AWPSO模型。对于40个硼原子的团簇(B40),他们找到了一个类平面结构,中间有两个六边形空位,原子排列成类正六边形结构,这相比B45团簇拥有更高的对称性和稳定性,最低能量值为-101954.52。
总结起来,这篇论文展示了如何利用机器学习和优化算法有效预测和寻找团簇的全局最优结构,为理解和设计新型材料提供了新的工具和方法。

第 5 页 共 29 页
了给定原子个数
n
以及所有原子两两间的相互距离
ij
r
外,还可利用对
的不同取值来表
示元素种类,另外
0
r
表示原子间的平衡距离。
3.
Sutto n Chen−
势能模型
Sutto n Chen−
势能模型常用于表示金属原子间的相互作用关系,由 A.P. Sutton 与 J.
Chen 于 1990 年提出,他们将势能表示为如下
Finnis Sinclair−
形式:
1
()
2
( ) ( )
()
T ij i
i j i i
n
m
i
ji
ij
E V r c
V r a r
a
r
= −
=
=
(3)
其中
c
为一个无量纲的正参数,
为能量单位参数,
a
为长度控制单位,
,mn
是正整数。
和
a
通常情况下取 1,
c
与
,mn
共同确定团簇中的原子种类,对于
Au
原子而言,
c
取 1,
,mn
分别为 8 和 10。
4.
Gupta
势能模型
( )
1
(0)
1( )
1/2
2
(0)
1( )
( ) ( )
( ) exp 1
( ) exp 2 1
n
rm
i
ij
r
ij ij
j j i
ij
ij
m
n
ij i
ji
n
j
j
j
n
i
V V i V i
r
V i A p
r
r
V i q
r
=
=
=
=−
= − −
= − −
(4)
Gupta
势能模型的精度相对较高,该模型也常用于描述金属原子间的相互关系,
(0)
ij
r
表示平衡距离,
, , ,
ij ij ij ij
A p q
共同确定团簇中的原子种类。由文献可知,针对
Au
原子的模
型非线性回归参数可选为:
0.2061
ij
A =
,
1.79
ij
=
,
10.229
ij
p =
,
4.036
ij
q =
,
(0)
1.0
ij
r =
。
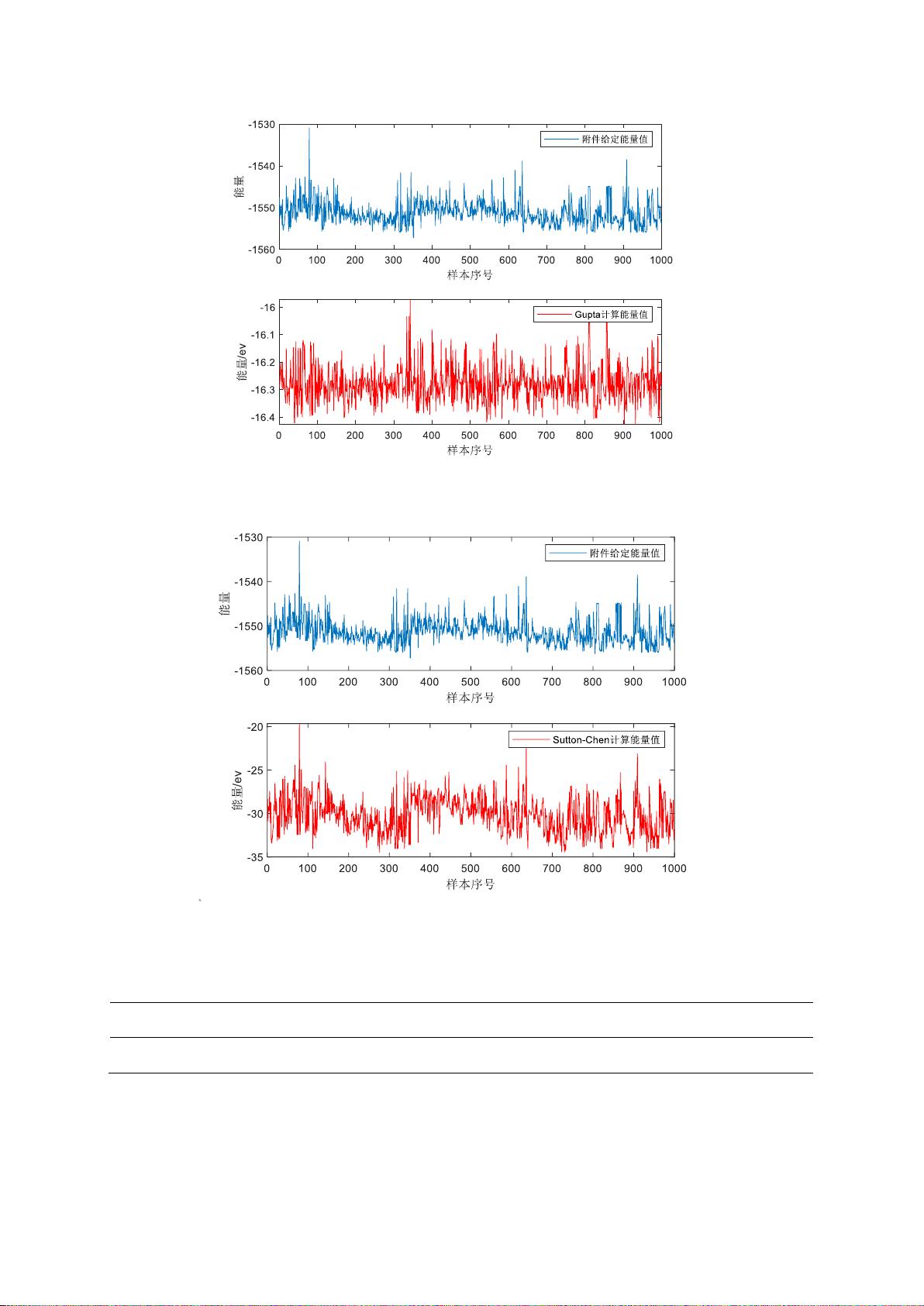
4.2.2 传统势能模型相关性分析
在建立四种常用模型的基础上,将给定的 1000 个
团簇样本数据分别带入模型
进行验证,得到每种模型对应的计算势能值。以
Gupta
与
Sutto n Chen−
势能模型为例,得
到的计算结果如图 4.1,图 4.2 所示。将通过各模型得到的计算势能值与真实值分别进行
相关性分析,可发现由
Sutto n Chen−
势能模型计算得到的能量值与实际样本中对应的能量
值呈现出较强的相关性,进一步对对实际与计算能量值进行非线性回归得到的拟合优度
如表 4.1 所示,对应散点图如图 4.3 所示,因此最终选择
Sutto n Chen−
势能模型作为构建
团簇能量预测模型的基础。
剩余43页未读,继续阅读
相关推荐
2022-08-08 上传
2022-08-03 上传
166 浏览量
327 浏览量
2022-10-13 上传
1246 浏览量
2024-05-03 上传
124 浏览量

巧笑倩兮Evelina
- 粉丝: 27
 我的内容管理
展开
我的内容管理
展开








最新资源
- Bmob云SDK 3.5.9版本发布:专为聊天工具开发
- AForge.NET框架许可证与依赖信息解析
- Html5图片上传实现及手机版兼容性分析
- exe4j最新版:Java转exe工具全面支持JDK1.7至1.8
- k-means算法实现与下载学习资源分享
- 下载Windows平台的.NET安装包教程
- VS2005/2008下打开VC6.0工程所需的LIBCDlib库下载
- 基于JavaFX开发的多功能数独游戏介绍
- Java版坦克大战小游戏发布:加入超级炮弹与机器人
- 全面解析WebQuestions数据集:问答系统的关键信息源
- 掌握SQL语句最优化技巧——《Effective MySQL》
- websocket在线聊天工具2.0:支持附件和涂鸦表情
- Linux下TCP聊天程序实现与文件传输功能探索
- SSM框架一键生成java实体类与MyBatis映射文件
- jQuery基础教程第四版:全面学习指南
- Git V1.9.5版本工具发布介绍
