利用异构网络中互信息的标签传播预测药物-靶标相互作用
38 浏览量
更新于2024-08-29
收藏 1.55MB PDF 举报
"这篇研究论文探讨了一种利用异质网络中的相互作用信息进行药物靶标交互预测的新方法——LPMIHN(Label Propagation with Mutual Interaction Information derived from Heterogeneous Networks)。"
在药物研发领域,预测药物与目标蛋白的相互作用(Drug-Target Interactions, DTIs)是一个关键任务,它对于理解蛋白质功能、提高药物研发效率特别是药物再定位具有重要意义。传统的药物发现过程是耗时且昂贵的,因此,发展有效的计算方法来预测DTIs已成为当前研究的热点。
近年来,已经提出了许多基于机器学习的算法来解决这个问题,例如基于核方法、矩阵分解方法以及网络推理方法等。这些算法的核心假设是相似的药物倾向于与相似的蛋白质相互作用,从而提高了预测药物和目标蛋白之间相互作用的准确性。然而,这些方法往往忽略了网络中丰富的异质信息,如药物化学结构、蛋白质功能、基因表达数据等。
LPMIHN方法正是为了解决这一问题而设计的。它通过构建一个包含多种类型节点和边的异质网络,如药物节点、靶标节点、药物化学结构节点、蛋白质功能节点等,来整合多元信息。然后,该方法利用标签传播策略,考虑了网络中节点之间的相互作用信息,来传播已知的DTI标签,从而预测未知的交互关系。这种方法能够更准确地捕捉到药物和靶标之间的复杂关系,提高预测的精确度。
在LPMIHN中,"相互作用信息"是指在异质网络中不同类型的节点之间存在的相互依赖和影响,这包括药物之间的化学相似性、蛋白质的生物学功能相关性等。通过对这些信息的量化和整合,LPMIHN能够更全面地描述药物和靶标的相互作用模式,从而提升预测的性能。
论文《通过异质网络中的相互作用信息进行标签传播预测药物靶标交互》详细阐述了LPMIHN的理论基础、算法实现步骤以及性能评估。作者通过对比实验验证了LPMIHN在预测DTIs方面的优越性,并讨论了其潜在的应用前景,为药物研发提供了一种新的计算工具。
这篇研究论文对药物研发领域有着重要的贡献,它不仅丰富了预测DTIs的方法论,还为后续的研究提供了新的思路,有望加速新药发现和现有药物的再利用。
522 | Mol. BioSyst., 2016, 12, 520--531 This journal is
©
The Royal Society of Chemistry 2016
heterogeneous network. Thirdly, implementation of label pro-
pagation on the drug (or target) similarity sub-network to
obtain the drug (or target) label network. Fourthly, implemen-
tation of label propagation on the target (or drug) similarity
sub-network, whose initial label information is derived from
the drug (or target) label network and the drug–target bipartite
network. Finally, the most probable targets (or drugs) are
selected according to the stable label scores of the walk.
LPMIHN is mainly different from NRWRH in three aspects.
One is that the drug/target similarity network integrates the
topological information of the known drug–target interaction
network. Another is that label propagation (or random walk) is
implemented on the drug and target similarity networks,
respectively. Thirdly, the initial label information of the target/
drug network comes from the drug/target label network and the
known drug–target bipartite network.
Through extensive simulations on four benchmark datasets
and two quantitative kinase bioactivity datasets, LPMIHN
shows better performance than the existing state-of-the-art
methods, such as BLM-NII, NetCBP and NRWRH. Furthermore,
some new predicted drug–target interactions ranked in top
were reported by publicly accessible datasets. It is anticipated
that our LPMIHN algorithm can help us to find new or potential
drug–target interactions, and provide useful information for
drug design.
2 Materials
To facilitate benchmarking comparison with other state-of-art
methods, we used the four drug–target interaction datasets
from humans, namely enzymes (Es), ion channels (ICs),
G-protein coupled receptors (GPCRs) and nuclear receptors
(NRs), which were originally provided by Yamanishi et al.,
40
and widely used as the benchmark binary interaction datasets
of compounds targeting pharmaceutically useful target pro-
teins.
29,31,34,35,42–44,47,48
These datasets are available at http://
web.kuicr.kyoto-u.ac.jp/supp/yoshi/drugtarget/. The Es dataset
includes 445 drugs, 664 targets and 2926 known drug–target
interactions. The ICs dataset includes 210 drugs, 204 targets
and 1476 known drug–target interactions. The GPCRs dataset
includes 223 drugs, 95 targets and 635 known drug–target
interactions. The NRs dataset includes 54 drugs, 26 targets
and 90 known drug–target interactions.
As binary interaction datasets ignore many important
characteristics of the drug–target interaction, such as dose-
dependence and quantitative affinity, we use the same cutoff
thresholds of K
d
r 30.00 nM and K
i
o 28.18 nM as ref. 18 to
binarize two large-scale quantitative kinase bioactivity datasets,
i.e., kinase disassociation constant (K
d
) dataset and kinase
inhibition constant (K
i
) dataset,
49,50
forming two binary inter-
action datasets which include 68 drugs, 442 targets and 1527
drug–target interactions for the K
d
dataset, and 1421 drugs, 156
targets and 3200 drug–target interactions for the K
i
dataset.
These two datasets are applied to evaluate the performance of
our LPMIHN algorithm. The smaller the K
d
/K
i
bioactivity, the
higher the interaction affinity between the chemical compound
and the protein kinase.
Table 1 lists some statistics of each dataset including the
total number of drugs (N
d
), the total number of targets (N
t
), the
total number of interaction edges (E
dt
), the total number of
drugs that have only one targeting protein (k
d
(1)), the total
number of targets that have only one associated drug (k
t
(1)), the
average number of targets for each drug (avg. N
d
), the average
number of drugs for each target (avg. N
t
), and the sparsity
which is defined as the total number of connected edges in the
real network divided by the total number of linked edges in the
complete graph.
3 Methods
Our LPMIHN method can be divided into two parts: construct-
ing the heterogeneous network and separately implementing
label propagation on the drug/target similarity networks.
3.1 Heterogeneous network
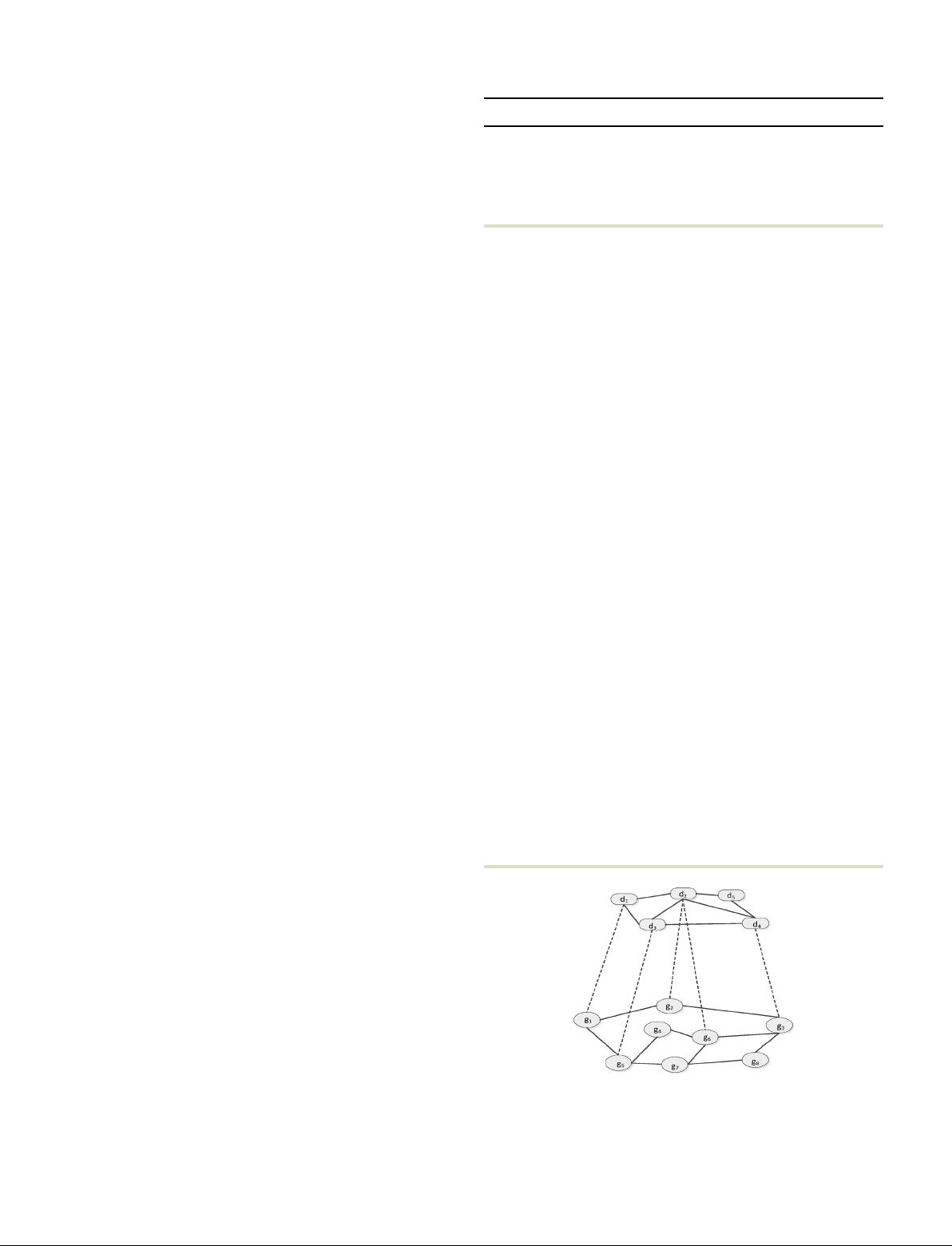
The heterogeneous network of drug–target interactions is
composed of three typical networks: the drug similarity net-
work, target similarity network and the known drug–target
interaction bipartite graph network (see Fig. 1).
The matrix S
d
corresponding to the drug similarity network
is composed of the chemical structure similarity matrix S
c
d
and
the drug–target interaction profile-based drug similarity matrix
S
IP
d
. The matrix S
g
corresponding to the target protein similarity
network is composed of the protein sequence similarity matrix
S
s
g
and the drug–target interaction profile-based target similarity
matrix S
IP
g
. The drug–target interaction adjacent matrix A
Table 1 Statistical characteristics of six drug–target interaction datasets
Dataset N
d
N
t
E
dt
k
d
(1) k
t
(1) avg. N
d
avg. N
t
Sparsity
Es 445 664 2926 177 288 6.58 4.41 0.0099
ICs 210 204 1476 81 23 7.03 7.24 0.0344
GPCRs 223 95 635 106 34 2.85 6.68 0.0299
NRs 54 26 90 39 8 1.67 3.46 0.0641
K
d
68 442 1527 4 97 22.46 3.45 0.0508
K
i
1421 156 3200 204 11 2.25 20.51 0.0144
Fig. 1 Drug–target interaction heterogeneous network model. The upper
sub-network is the drug similarity network, the underlying sub-network is
the target protein similarity network and the intermediat e layer is a drug–
target interaction bipartite graph network.
Molecular BioSystems Paper
剩余11页未读,继续阅读
2020-03-20 上传
2022-06-02 上传
2021-04-13 上传
2021-03-21 上传
2021-04-24 上传
2021-08-04 上传
2021-04-06 上传
2018-09-19 上传
2021-06-29 上传

weixin_38721652
- 粉丝: 3
- 资源: 935
 我的内容管理
展开
我的内容管理
展开







最新资源
- hackerrank 30天挑战
- SMStagger:文字排程应用程式
- rick-morty-app-chpx
- Java_script_slide-show
- events-app-angular
- ECMO-Device-Simulation
- showdialog010220
- LinuxJava(TM) SE 1.8 and MysqlJava
- randomAnimalGenerator:阿基德阿基特图拉-德阿皮
- portafolioWeb:网络作品集项目
- SocialTab-crx插件
- 转子动力学工具箱 (RotFE):工具箱对带圆盘的旋转弹性轴进行建模-matlab开发
- robinlennox.github.io
- 异构数据库迁移同步(搬家)工具.zip
- Accuinsight-1.0.18-py2.py3-none-any.whl.zip
- Unity:Unity脚本
