Materials Studio入门:分子建模与量子计算详解
需积分: 14 52 浏览量
更新于2024-08-02
收藏 679KB PDF 举报
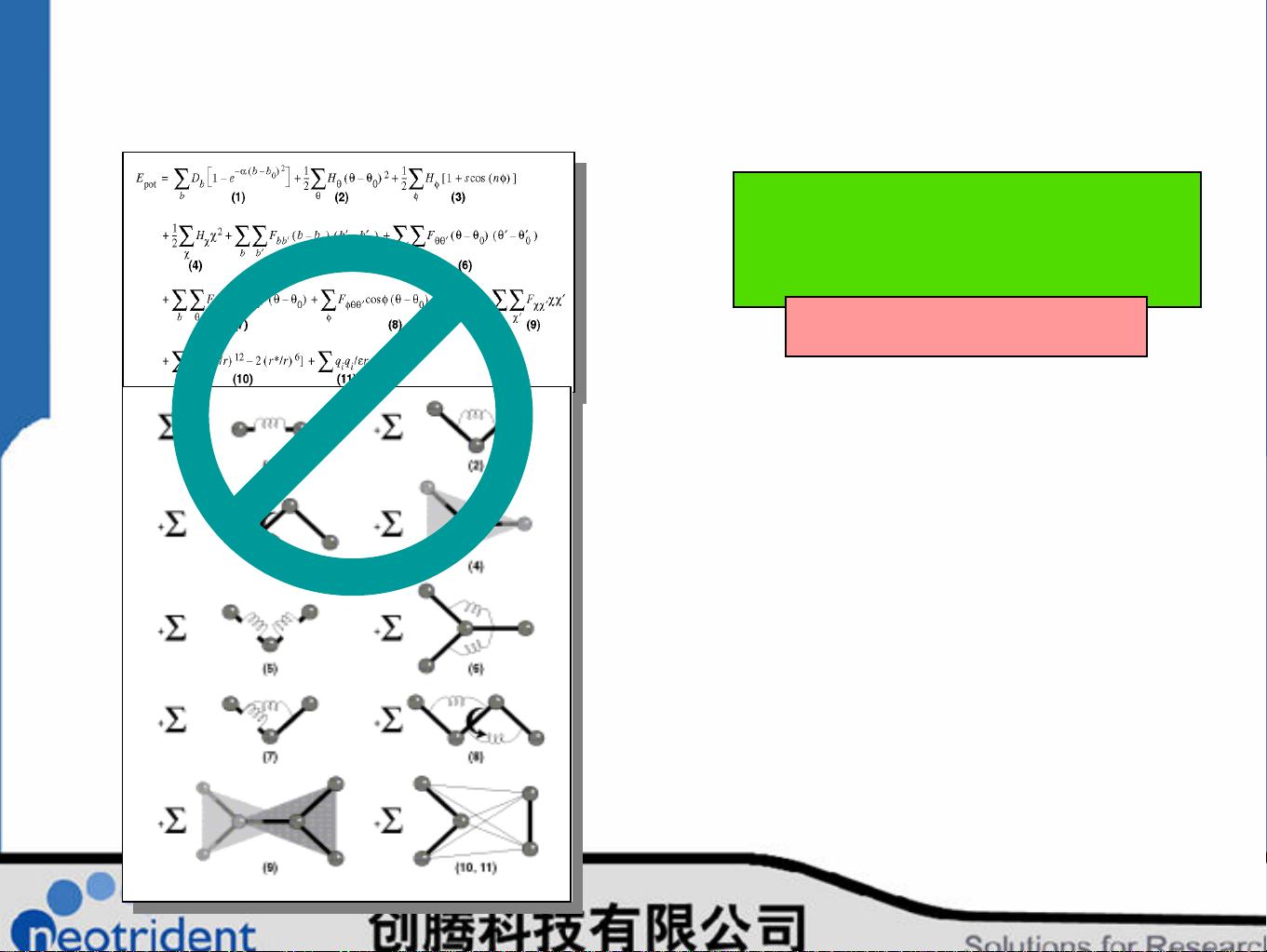
"Materials Studio初级入门教程深入探讨了材料分子建模计算的重要性和应用。这是一份针对初学者的指南,旨在引导读者掌握这款强大的软件,包括Visualizer工具在聚合物、有机分子以及表面和晶体模拟中的运用。它特别提到了VAMP(Virtual Atomic Manipulation Platform),这是一个在个人电脑上进行半经验模拟的技术,提供了快速的结构优化和性质预测功能。
在课程中,重点讲解了Premier Density Functional Theory (DFT)程序,如CASTEP,它是用于模拟固体、界面、表面特性的主要工具。例如,通过例子介绍了chemisorption(如CO在Pd表面的吸附)和气体(如CH4在Ni(111)表面的解离)过程,这些是理解材料表面化学反应的关键步骤。
DFT与量子力学(QM)相结合,比如HΨ=EΨ这一基本原理,表明通过哈密顿算符作用于波函数,可以得到系统的性质。虽然力场方法能够提供合理的结构和构象分析,但在精确研究过渡态时,更复杂的量化方法如Hartree、Fock、CI(Configuration Interaction)、TB( Tight Binding)等理论和方法显得尤为重要,因为它们无需实验参数,适用于所有元素,并且能够处理键的断裂等问题。
此外,材料科学家们经常使用的DMol3是一种量子力学方法,它通过解决薛定谔方程来处理原子间相互作用,这是计算化学的基础。Hartree-Fock和密度泛函理论(DFT)作为两种常见的近似方法,分别基于独立电子和平均场的概念,而CI则提供了更准确但计算成本更高的多电子相互作用处理。
本教程涵盖了从基础概念到高级应用的全面内容,帮助用户理解为何需要量化手段,以及如何在Materials Studio这个平台上高效地进行结构模拟和性质预测,无论是处理简单的聚合物还是复杂的化学反应,都是宝贵的资源。"
H
Ψ
= E
Ψ
E. Schrödinger, 1926
为什么我们需要量化手段?
对于结构和构象,力场方法能
够给出相当合理的结果……
但是如果要精确的测定过渡态,则
需要更复杂的量化方法……
•
•
没有实验参数
没有实验参数
•
•
处理所有的元素
处理所有的元素
•
•
键的断裂
键的断裂
…

剩余42页未读,继续阅读
2020-04-13 上传
2020-04-13 上传
2023-11-11 上传
2021-09-19 上传
2021-09-30 上传
2023-09-13 上传
2022-12-25 上传

jiangxiabing
- 粉丝: 1
- 资源: 1
 我的内容管理
展开
我的内容管理
展开







最新资源
- Cucumber-JVM模板项目快速入门教程
- ECharts打造公司组织架构可视化展示
- DC Water Alerts 数据开放平台介绍
- 图形化编程打造智能家居控制系统
- 个人网站构建:使用CSS实现风格化布局
- 使用CANBUS控制LED灯柱颜色的Matlab代码实现
- ACTCMS管理系统安装与更新教程
- 快速查看IP地址及地理位置信息的View My IP插件
- Pandas库助力数据分析与编程效率提升
- Python实现k均值聚类音乐数据可视化分析
- formdotcom打造高效网络表单解决方案
- 仿京东套餐购买列表源码DYCPackage解析
- 开源管理工具orgParty:面向PartySur的多功能应用程序
- Flutter时间跟踪应用Time_tracker入门教程
- AngularJS实现自定义滑动项目及动作指南
- 掌握C++编译时打印:compile-time-printer的使用与原理
