Lammps在分子动力学模拟中的应用与优势
需积分: 30 47 浏览量
更新于2024-07-20
1
收藏 320KB PPT 举报
"分子动力学模拟是通过数值方法研究物质微观结构和动态行为的重要手段,而LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)是一个广泛使用的开源分子动力学模拟软件。本资料主要讨论了LAMMPS在分子动力学模拟中的应用,并与其他知名分子动力学程序进行了对比。此外,还介绍了使用Lennard-Jones(L-J)势模拟裂纹扩展的相关内容。"
分子动力学模拟是一种基于牛顿运动定律的方法,用于追踪大量粒子在时间上的运动轨迹,以研究物质的微观行为。这一领域中有多个著名的分子动力学模拟程序,包括NAMD、AMBER、CHARMM、GROMACS、TINKER、DL-POLY以及LAMMPS。这些程序各有特点,适用于不同的研究领域。
NAMD是一款专为生物和化学软材料体系设计的免费软件,以其高效能和强大的VMD分析工具而闻名。AMBER则主要针对生物体系,提供丰富的内置势能模型,易于自定义新的分子模型。CHARMM同样关注生物体系,具有快速更新的势能模型。GROMACS以其高效的算法和友好的用户界面在生物体系模拟中表现出色。TINKER是一个通用软件,略侧重于生物体系,支持多种模型,但还在持续发展中。DL-POLY是一款界面友好的软件,适用于各种分子模拟,计算效率较高。
LAMMPS作为一款免费的一般性分子模拟软件,不仅兼容多种势能模型,而且编程水平高,计算效率出色,能够处理软材料和固体物理系统。MaterialsExplorer是专为Windows设计的多功能分子动力学软件,涵盖广泛的应用领域,如有机物、高聚物、生物大分子等,并提供了丰富的力场选择。
在使用L-J势模拟裂纹扩展时,Lennard-Jones势是描述分子间相互作用的经典模型,通常需要进行无量纲化处理,以便在不同规模和条件下进行计算。无量纲化的单位系统简化了计算,使模拟更具普适性。L-J势可以有效地模拟原子间的吸引和排斥作用,对于理解材料的裂纹形成和扩展过程至关重要。
通过LAMMPS和其他分子动力学软件的对比,研究者可以根据具体需求选择最适合的工具,进行各种复杂系统的模拟,例如理解材料的机械性能、相变、热力学性质等。同时,掌握L-J势的使用可以帮助科学家深入探究材料的断裂行为,这对于材料科学和工程领域的研究具有重要价值。
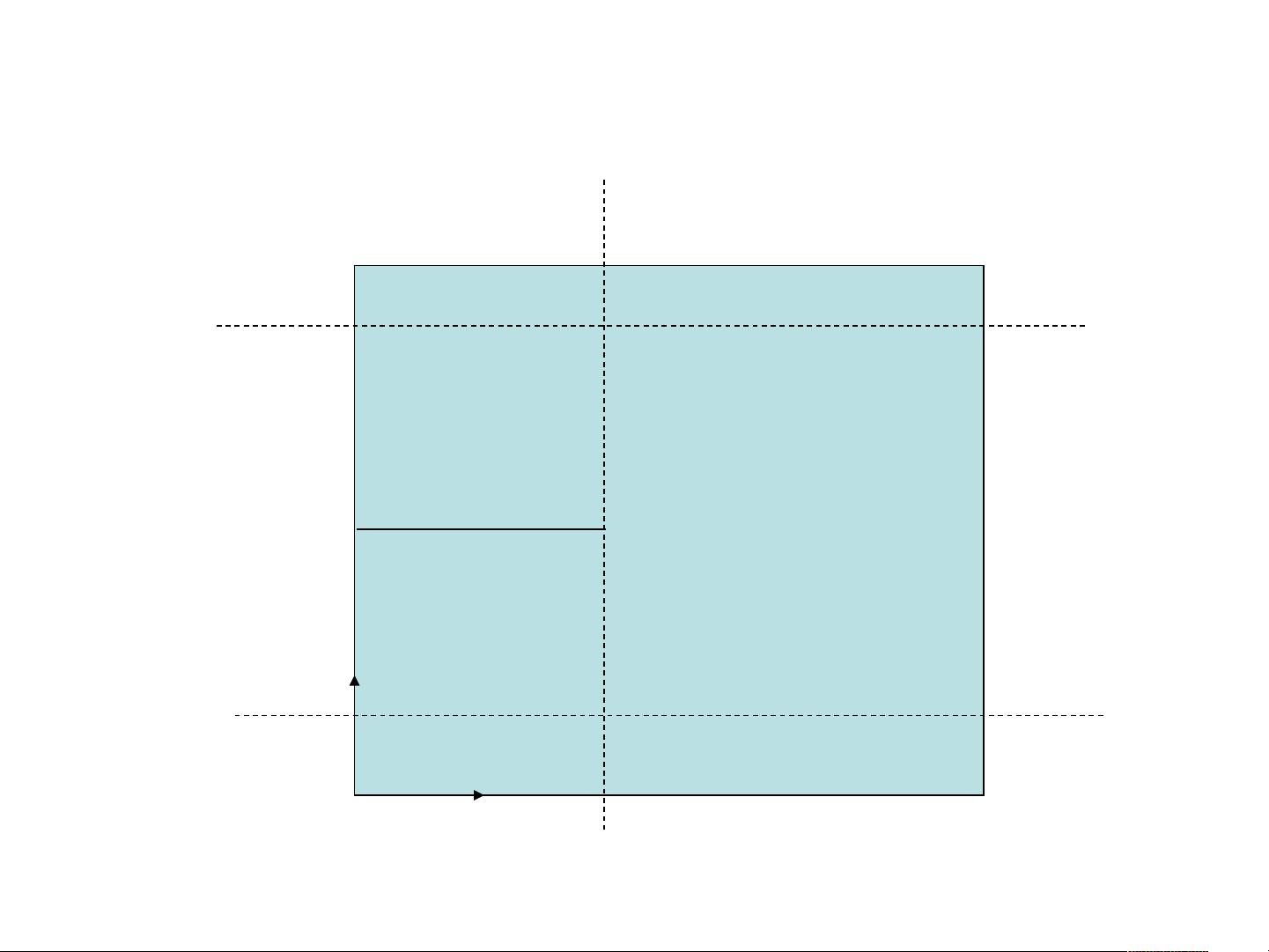
B 、使用 L-J 势模拟裂纹的扩展
裂纹
lower
upper
leftupper
leftlower
x
y

剩余32页未读,继续阅读
点击了解资源详情
点击了解资源详情
点击了解资源详情
2022-07-14 上传
116 浏览量
109 浏览量
2021-09-10 上传
2019-01-20 上传

sinat_36457457
- 粉丝: 0
- 资源: 1
 我的内容管理
展开
我的内容管理
展开







最新资源
- MATLAB新功能:Multi-frame ViewRGB制作彩色图阴影
- XKCD Substitutions 3-crx插件:创新的网页文字替换工具
- Python实现8位等离子效果开源项目plasma.py解读
- 维护商店移动应用:基于PhoneGap的移动API应用
- Laravel-Admin的Redis Manager扩展使用教程
- Jekyll代理主题使用指南及文件结构解析
- cPanel中PHP多版本插件的安装与配置指南
- 深入探讨React和Typescript在Alias kopio游戏中的应用
- node.js OSC服务器实现:Gibber消息转换技术解析
- 体验最新升级版的mdbootstrap pro 6.1.0组件库
- 超市盘点过机系统实现与delphi应用
- Boogle: 探索 Python 编程的 Boggle 仿制品
- C++实现的Physics2D简易2D物理模拟
- 傅里叶级数在分数阶微分积分计算中的应用与实现
- Windows Phone与PhoneGap应用隔离存储文件访问方法
- iso8601-interval-recurrence:掌握ISO8601日期范围与重复间隔检查
