新型基因组组装算法SWA:高效处理重复与非重复序列
71 浏览量
更新于2024-07-14
收藏 1.59MB PDF 举报
本文主要探讨了"重复和非重复的从头基因组装配算法"这一研究主题。随着新一代测序技术(Next-Generation Sequencing, NGS)的发展,其产生的短读长度、高覆盖和高通量特性使得在进行特定基因组分析前进行从头组装成为可能。然而,当前的基因组组装方法在处理重复序列时存在显著性能问题。针对这一挑战,研究人员提出了名为SWA(Short-read Whole-genome Assembler)的新算法。
SWA算法具有四个关键特性:首先,它能够同时组装重复和非重复序列,这是传统方法往往难以兼顾的;其次,通过引入新的重叠扩展策略,每个种子区域的扩展更加有效,提高了组装效率;第三,SWA采用了滑动窗口滤除排序偏差的技术,以减少由测序数据排序不一致带来的影响;最后,针对低覆盖率数据集,算法还设计了一种补偿机制,增强了在数据稀疏情况下的表现。
在实验评估中,SWA展示了出色的性能。在仿真数据集和真实测序数据上,重复组装的准确率高达99%,而估计的拷贝数准确性更是达到了100%。这表明SWA在处理重复序列时的精确度非常高。此外,与市面上其他八家领先的基因组组装工具进行对比,SWA在重复和非重复序列的完整性以及正确性方面表现出明显优势。
本文提出的SWA算法革新了从头基因组组装方法,特别在应对复杂重复序列时,不仅能够准确地定位重复和非重复序列,还能实现它们的完整组装,这使得它在同类工具中脱颖而出。这对于基因组学研究中的高质量数据分析具有重要意义,尤其是在那些依赖于重复序列信息的领域,如基因家族分析和遗传变异研究。SWA的开源许可模式也使得该算法易于获取和应用,进一步推动了基因组学领域的技术进步。
BioMed Research International
A
NN
AAAAAA
C
G
T
Sorted reads
Discard reads
Raw reads
(a)
Processed
5
Frequency
3
8
3
4
raw reads
Unique reads
(b)
Frequency
Read count
Seeds of repeats
Seeds of non-repeats
(c)
Seed
Seed
1.3 1.3 2 2 2.3 1.6 1.3 1.3
C1
C2
1
2
1322121
1
2
1322121
Dynamic overlap
interval
r
1
r
3
r
2
Read counts in dynamic
overlap interval
Filtering by sliding
window
Filtered by
sliding window
(d)
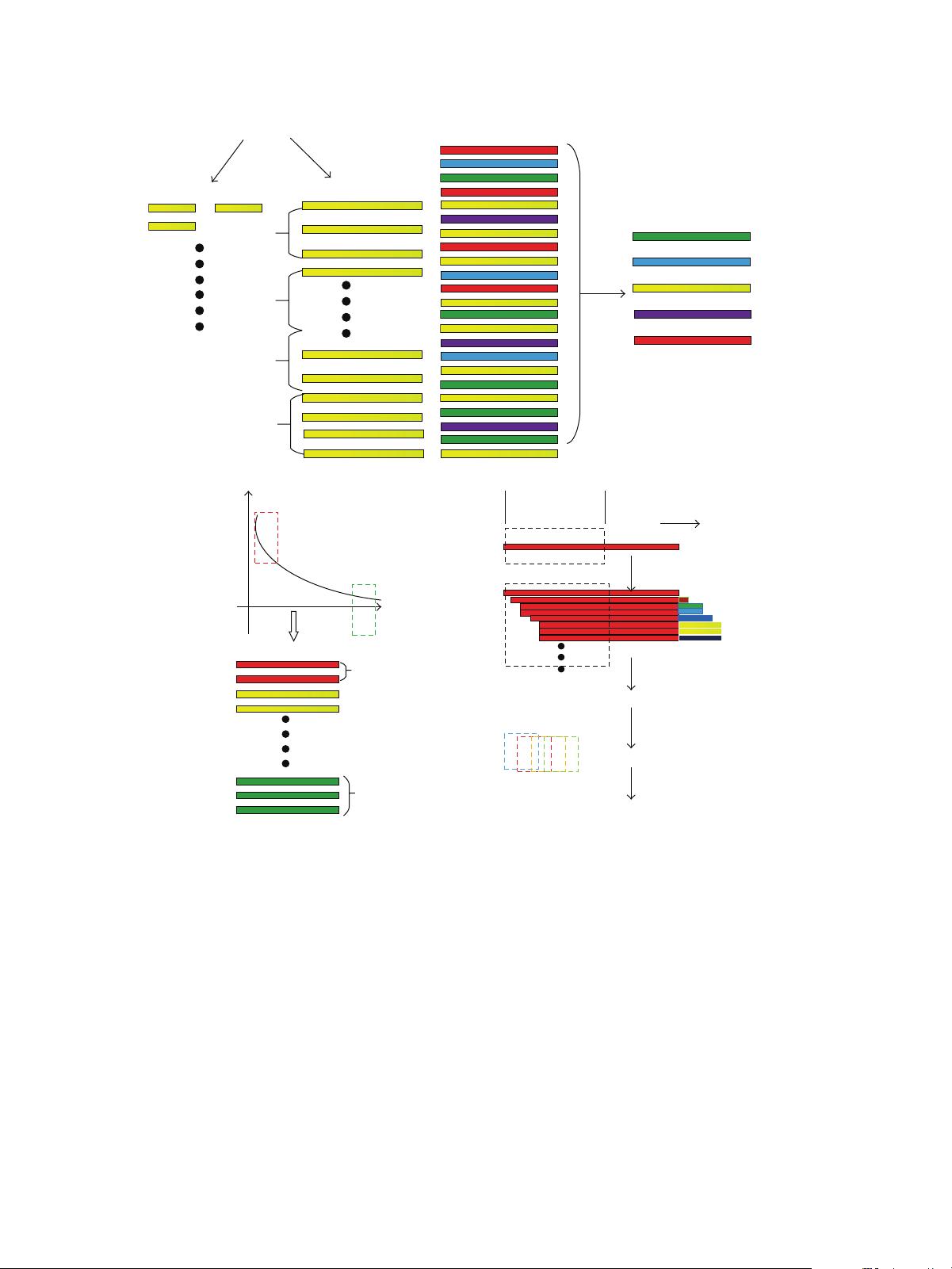
F : Some key steps of SWA. (a) Raw reads processing. Input reads containing any “𝑁”oralowqualityregionarediscardedandthen
sorted in alphabetical order. (b) Graphical illustration of unique process. e ve dierent color lines represent the ve unique reads in
preprocessed raw reads. Each of them appears more than twice. By unique processing, the identical reads are collapsed into one unique and
corresponding frequency. (c) Seed selection. e unique reads are ranked by read count (from high to low). Unique reads with read count
larger than 𝐻
𝑝
are selected as seeds for repeat (the red dotted frame), while unique reads with read counts smaller than 𝐿
𝑝
are selected as seeds
for nonrepeat extension (the blue dotted frame). (d) e graphical example of extending repeats using sliding window function in dynamic
overlapping interval. e dotted box represents the dynamic overlapping interval 𝐿
𝑑
. Aer overlapping with seed in 𝐿
𝑑
, the overlapped read
counts are recorded and then sliding window function is used to lter out the read bias in this interval continuously, as shown in C. C is the
corresponding results ltered by sliding window function, and then the mean value of this interval is recorded in variable 𝑀
𝑛
to detect the
boundary of repeats (Figure ) and nonrepeats (Figure ). In this extension, SWA regards 𝑟
1
as the optimal extendable read. e extension of
nonrepeats is performed in a similar way. e detailed extension and boundary detection are graphically shown in Figures and .
Fourthly, seed selection is conducted. In SWA, each
extension requires a unique read, called a seed, to initiate
the extension. In an extension-based assembler, a good seed
should not contain any sequencing errors and should not
be selected from the boundary of repeats and nonrepeats.
Consequently, data cleaning is necessary in the data
preprocessing stage before seed selection, and then the seeds
areselectedintable𝑅. In addition, theoretically, a read
from repetitive region usually has a high read count because
identical repeats from other loci are counted as well; a read
剩余16页未读,继续阅读
2018-07-13 上传
2022-06-10 上传
2021-05-16 上传
2021-05-24 上传
2021-05-27 上传
2021-05-03 上传
2021-05-05 上传
2021-04-12 上传
2021-03-18 上传

weixin_38577200
- 粉丝: 9
- 资源: 907
 我的内容管理
展开
我的内容管理
展开







最新资源
- 高清艺术文字图标资源,PNG和ICO格式免费下载
- mui框架HTML5应用界面组件使用示例教程
- Vue.js开发利器:chrome-vue-devtools插件解析
- 掌握ElectronBrowserJS:打造跨平台电子应用
- 前端导师教程:构建与部署社交证明页面
- Java多线程与线程安全在断点续传中的实现
- 免Root一键卸载安卓预装应用教程
- 易语言实现高级表格滚动条完美控制技巧
- 超声波测距尺的源码实现
- 数据可视化与交互:构建易用的数据界面
- 实现Discourse外聘回复自动标记的简易插件
- 链表的头插法与尾插法实现及长度计算
- Playwright与Typescript及Mocha集成:自动化UI测试实践指南
- 128x128像素线性工具图标下载集合
- 易语言安装包程序增强版:智能导入与重复库过滤
- 利用AJAX与Spotify API在Google地图中探索世界音乐排行榜
