LAMMPS分子动力学模拟教程:从入门到进阶
需积分: 0 56 浏览量
更新于2024-07-03
10
收藏 1.92MB PDF 举报
"分子动力学模拟及其LAMMPS实现-讲义.pdf"
这是一份关于分子动力学模拟的讲义,特别关注了LAMMPS软件的使用和实现。分子动力学模拟是一种强大的计算方法,用于研究物质在原子或分子级别的动态行为。LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)是分子动力学领域广泛应用的开源软件,适用于模拟各种物理和化学过程。
1. 分子动力学基础:
- 原理:分子动力学基于牛顿运动定律,通过数值求解来追踪每一个分子的运动轨迹,从而揭示系统的宏观性质。
- 势能:分子间相互作用通常由势能函数描述,如Lennard-Jones势、库仑势等,这些势能函数决定了分子间的吸引力和排斥力。
- 温度、压强:分子动力学模拟可以计算系统的温度和压强,它们是通过分析分子的动能分布和体积变化来确定的。
2. Linux系统常见命令和操作:
- 在进行LAMMPS模拟时,通常需要在Linux环境下工作,因此了解基本的Linux命令和操作是必要的,如文件管理、编译、运行程序等。
3. LAMMPS介绍和并行版安装(Linux版):
- LAMMPS提供了丰富的功能,包括各种分子间相互作用势、并行计算支持、多种模拟算法等。
- 安装过程涉及编译源代码,配置并行环境,例如使用MPI(Message Passing Interface)进行多核或多节点并行计算。
4. LAMMPS模拟基本流程:
- 输入文件编写:定义系统的粒子类型、坐标、力场参数、时间步长等。
- 初始化系统:创建初始构型,设置温度、压强等初始条件。
- 运行模拟:执行分子动力学演化,记录关键数据。
- 后处理分析:利用输出数据进行统计分析,如结构、能量、动力学性质等。
5. 专题内容:
- 从纳米流动、纳米流体到润湿性,再到材料的拉伸、压缩、弯曲、扭转、剪切性能,以及传热、粘度、扩散、摩擦和磨损等,涵盖了广泛的物理现象。
- 反应分子动力学ReaxFF允许模拟化学反应过程,而耗散粒子动力学(DPD)则适用于软物质和生物系统。
- 自由能计算对于理解相变和化学平衡至关重要,原子沉积和镀膜模拟则应用于材料生长过程。
这份讲义适合对分子动力学和LAMMPS感兴趣的学生和研究人员,特别是初学者,旨在通过逐步指导降低学习门槛,使他们能够更快地掌握LAMMPS并专注于科学研究。讲义还鼓励社群交流,提供微信联系方式以便于互助学习和社区建设。
添加微信:baolu_yao,进入微信群,帮助他人,共建社区
10
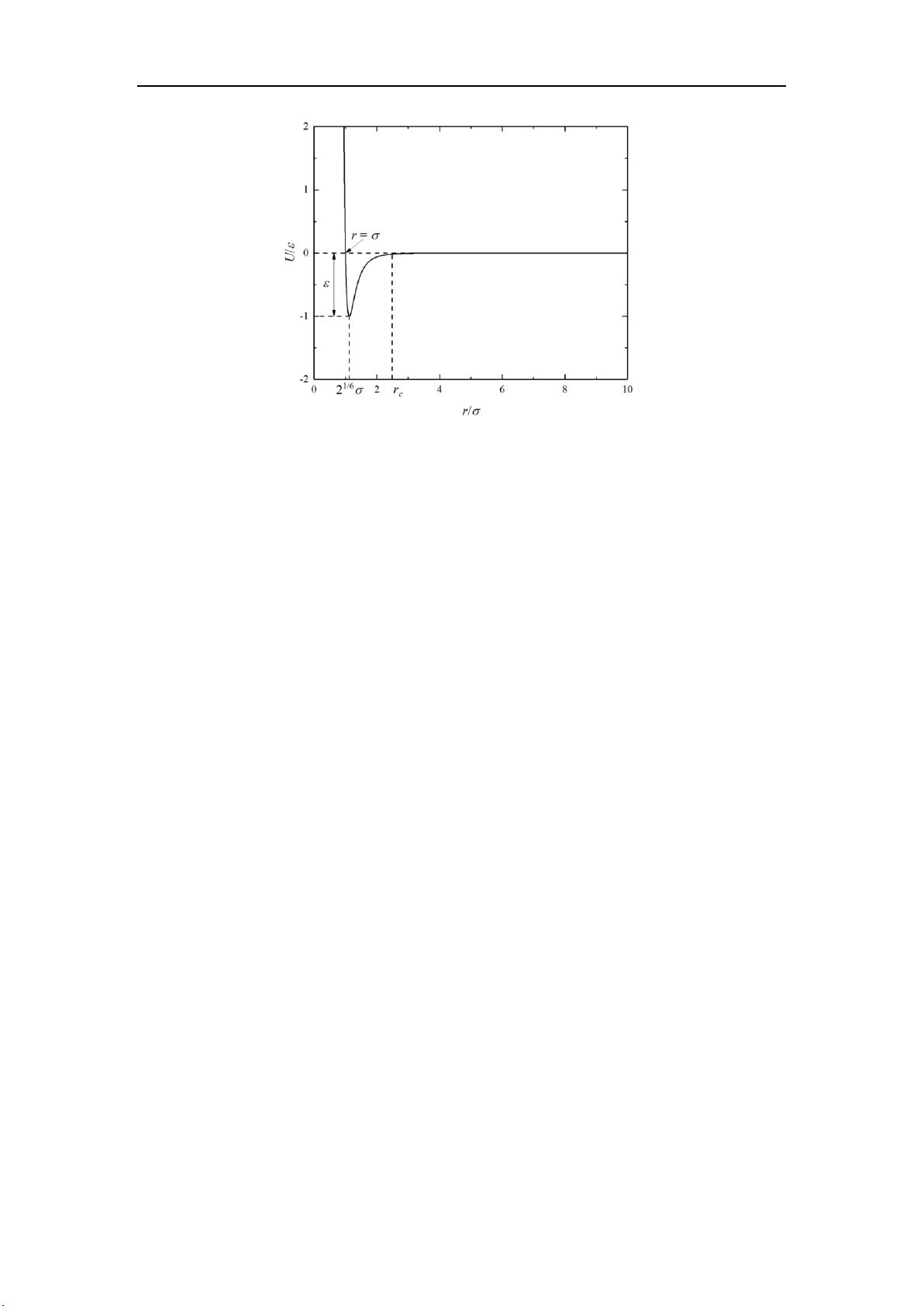
上图是 LJ/126 势函数的图像。对于该势函数有几个重要的点要说一下。势能的
最低点的到零点之间的绝对值为,称为势能函数的特征能量,势能的零点位于
处,称为势能函数的特征长度。势能函数的最低点位于
处,在该
点处势能对位置的导数是零,所以该点处受力也为零。该点也称为平衡位置,我
们可以看到在平衡位置出势能是负的。所以当体系处于平衡状态时,原子之间的
相对距离大概也在平衡位置附近,所以势能为负的。这也就是为什么大多数情况
分子动力学算出来的势能都是负的。在
,两个原子之间表现为相互排斥,
在
,两个原子之间表现为相互吸引。从势函数的图像上可以看出在较
大的时候,势能趋于零,其导数也趋于零。也就是当两个原子距离很远的时候,
原子之间的相互作用可以忽略不计。因此在实际计算的时候都会使用截断的
LJ/126。截断距离成为截断半径,其值大概在。就像重力势能一样,势能的
计算需要一个参考位置。LJ/126 的参考位置选择在了无限远处。类似 LJ/126 势
能以及其他拓展的 LJ 势能,都称为对势,其含义是两个原子之间的相互作用只
由这一对原子决定,与其它相邻原子没有关系。
分子物质
原子之间有化学键连接的物质。比如常见的水,甲烷,乙醇,醋酸,氯化钠,聚
乙烯,酯类,氨基酸,RNA,DNA 等。分子物质是分子动力学模拟应用最广的
对象之一。为了描述分子物质研究者开发出了一系列力场。值得说明的是力场是
一个经验性的东西。研究者通过自己的物理直觉和反复试验,最终建立起适合某
一类物质的力场。对于分子物质,相互作用包括两类:非键结势能和键结势能。
非键结用来描述没有化学键连接原子之间的相互作用,键结势能用来描述有化学

剩余59页未读,继续阅读
429 浏览量
584 浏览量
2022-03-09 上传
2022-04-13 上传
121 浏览量

分子模拟全能助手
- 粉丝: 122
- 资源: 1
 我的内容管理
展开
我的内容管理
展开







最新资源
- 软件水平考试网络工程师英语复习练习题10套
- JAVA面试题目大汇总
- 门禁系统设计 论文 完整版
- soa相关技术介绍与实现
- a Frame Layout Framework
- Thinking in Patterns
- 图书管理信息系统 SIM SQL Server2000数据库管理系统
- Bayesian and Markov chain
- Analysis of a Denial of Service Attack on TCP.
- 802.11英文原版协议 11G 11 N WEP WPA WPA2 BEACON 好东西大家分享
- aix双机配置详细配置
- 中国联通SGIP1.2
- 09数据库系统工程师考试大纲
- DFBlaser窄线宽激光器
- WinSock编程基础原理与C实现代码
- bfin-uclinux内核的CPLB v0.1
