神经诱导图卷积网络:miRNA-疾病关联预测的新方法
49 浏览量
更新于2024-08-27
收藏 1.27MB PDF 举报
本文是一篇发表于2022年的研究论文,标题为"Neural Inductive Matrix Completion with Graph Convolutional Networks for miRNA-disease Association Prediction",并收录于Bioinformatics杂志。该研究的动机在于快速预测miRNA(微小RNA)与疾病之间的关联,因为生物实验检测这种关联耗时且成本高。当前,计算机方法被广泛视为识别miRNA与疾病潜在关联的有效补充手段。
研究者们提出了一种新颖的方法——神经归纳矩阵完成与图卷积网络(Neural Inductive Matrix Completion with Graph Convolutional Networks, NIMCGCN),用于增强miRNA-disease关联的预测能力。NIMCGCN的核心原理是利用图卷积网络(GCNs)来学习miRNA和疾病之间的复杂关系。图卷积网络在生物信息学中应用广泛,因为它能够捕捉节点(miRNA和疾病)间的局部结构特征,并通过多层信息传递来整合全局信息。
论文的流程包括首先构建一个包含miRNA和疾病节点的图谱,其中边代表它们之间的相互作用或相似性。然后,NIMCGCN运用GCN进行特征提取,通过迭代更新节点的表示,以捕获其在图中的深层语义。这种方法允许模型在无监督或半监督的情况下学习到潜在的miRNA-disease关联模式,而无需大量的标注数据。
作者团队——来自云南大学软件学院的研究者们,包括Jin Li、Sai Zhang、Tao Liu、Chenxi Ning、Zhuoxuan Zhang和Wei Zhou——通过实验验证了NIMCGCN在预测准确性和效率上的优势,相比于传统的矩阵填充方法,NIMCGCN在miRNA-disease关联预测任务上取得了显著的性能提升。此外,文章还提到了接收日期、修订日期和接受日期,以及对应的联系人信息和审稿人姓名,这些都是论文发表过程中的关键细节。
这篇研究为miRNA-disease关联预测领域提供了创新的机器学习框架,有助于生物医学研究者更有效地预测潜在的疾病相关miRNA,从而推动个性化医疗和疾病预防的发展。
which is used to supplement the missing entries in MISIM. Specifi
cally,
mGS
𝑖,𝑗
is calculated by follows
mGS
𝑖,
𝑗
=exp
−𝜃
m
‖
𝐓
𝑖,:
−𝐓
𝑗
,:
‖
2
(2)
where 𝐓
𝑖,:
represents the 𝑖-row in the adjacent matrix 𝐓 and 𝜃
is the
kernel bandwidth parameter which is calculated by the following formula
𝜃
=
1
𝑚
‖
𝐓
𝑖,:
‖
(3)
where 𝑚 is the number of miRNAs i.e., the row number of 𝐓.
With
MS
𝑖,𝑗
, the miRNA functional similarity matrix is denoted by
𝐀
∈ℝ
𝑚×𝑚
and constructed by
𝐀
=MS
𝑖,𝑗
.
2.3 Disease-disease Similarity
The MeSH database (http://www.ncbi.nlm.nih.gov/) is available for
studying the relationship between different diseases. We obtained a hier-
archical directed acyclic graph (DAG) directly from MeSH, where each
node represents a disease and each directed edge in the DAG is from a
general disease term to a specific disease term.
The semantic similarity scores between different diseases were calcu-
lated based on disease DAG. First, let 𝑖∈D be a disease. dag
𝑖
indicates
the node set, including node 𝑖 and its ancestor nodes in the disease DAG.
Then, the first semantic contribution of a disease 𝑡∈D to the disease 𝑖 is
denoted by
SC
𝑖,
𝑡
and
can be formulated using the following equations
(Chen et al., 2018a),
SC
1
𝑖,𝑡
=1 𝑖𝑓𝑡=𝑖
SC
1
𝑖,𝑡
=max
𝛾 SC
1
𝑖,𝑡
|
𝑡
∈𝑐ℎ𝑖𝑙𝑑𝑟𝑒𝑛 𝑜𝑓 𝑡
𝑖𝑓 𝑡𝑖
(4)
where 𝛾 is a semantic contribution decay factor, which shows that as the
distances between disease 𝑡 and its ancestor diseases increases, their con-
tribution to the semantic value of disease 𝑑 progressively decreases. 𝛾 was
set as 0.5 according to previous literature (Wang et al.,2010).
Based on the definition of semantic contribution in Eq (4), the first
semantic similarity scores between different diseases, denoted by dS
was
established. Let 𝑖,𝑗 be two different diseases. dS
𝑖,𝑗
is defined as fol-
lows.
dS
𝑖,
𝑗
=
∑
SC
1
𝑖,𝑡
+ SC
1
𝑗
,𝑡
∈∩
∑
SC
1
𝑖,𝑡
𝑡∈
+
∑
SC
1
𝑗
,𝑡
𝑡∈dag
(5)
Intuitively, dS
𝑖,𝑗
is higher if the larger part of DAG is shared by i and
j.
However, dS
ignores the significance of different disease contribu-
tions. Supposing that 𝑖,𝑡,𝑞∈ D, if disease 𝑡 only appears in the dag
𝑖
,
and 𝑞 appears in both dag
𝑖
and the dag of other diseases, 𝑡 might have
higher semantic contribution to 𝑖 than 𝑞. Thus, the second semantic con-
tribution score SC
𝑖,𝑡
was presented as follows:
SC
𝑖,𝑡
=−log
the number of dags including 𝑡
the number of disease
(6)
Based on SC
𝑖,𝑡
, the second semantic similarity score dS
, between two
diseases was presented as follows (Chen et al., 2018a)
dS
𝑖,
𝑗
=
∑
SC
2
𝑖,𝑡
+ SC
2
𝑗
,𝑡
∈∩
∑
SC
2
𝑖,𝑡
𝑡∈
+
∑
SC
2
𝑗
,𝑡
𝑡∈dag
(7)
As disease similarity measures calculated using dS
and dS
are both
from the MeSH database, it provides only a part of the entries in diseases
semantic similarity matrix. Hence, the Gaussian interaction profile kernel
similarity was adopted to complement the remaining disease similarity en-
tries.
Specifically, let 𝐓∈
0,1
×
be the adjacent matrix constructed us-
ing the known HMDD v2.0 miRNA-disease association data. 𝐓
:,𝑗
is the
𝑗-column binary vector representing disease 𝑗. Then, Gaussian interaction
profile kernel similarity between disease 𝑖 and disease 𝑗 is defined as
dGS
𝑖,
𝑗
=exp
−𝜃
‖
𝐓
:,𝑖
−𝐓
:,
𝑗
‖
(8)
where 𝜃
is the kernel bandwidth parameter calculated using the follow-
ing formula
𝜃
=
1
𝑛
‖
𝐓
:,
𝑗
‖
(9)
where 𝑛 is the number of diseases i.e., the column number of 𝐓.
With dS
, dS
,
and dGS, the disease semantic similarity matrix is de-
noted by
𝐀
∈ℝ
𝑛×𝑛
and constructed using
𝐀
=
dS
1
𝑖,
𝑗
+dS
2
𝑖,
𝑗
2
,if𝑖and
𝑗
has semantic similarity score
dGS
𝑖,
𝑗
, otherwise
(10)
2.4 miRNA-disease Heterogeneous Information Network
We combined the miRNA functional similarity network 𝐀
, disease
semantic similarity network 𝐀
, and experimentally valid miRNA-disease
interactions
𝐓
to obtain the whole miRNA-disease heterogeneous infor-
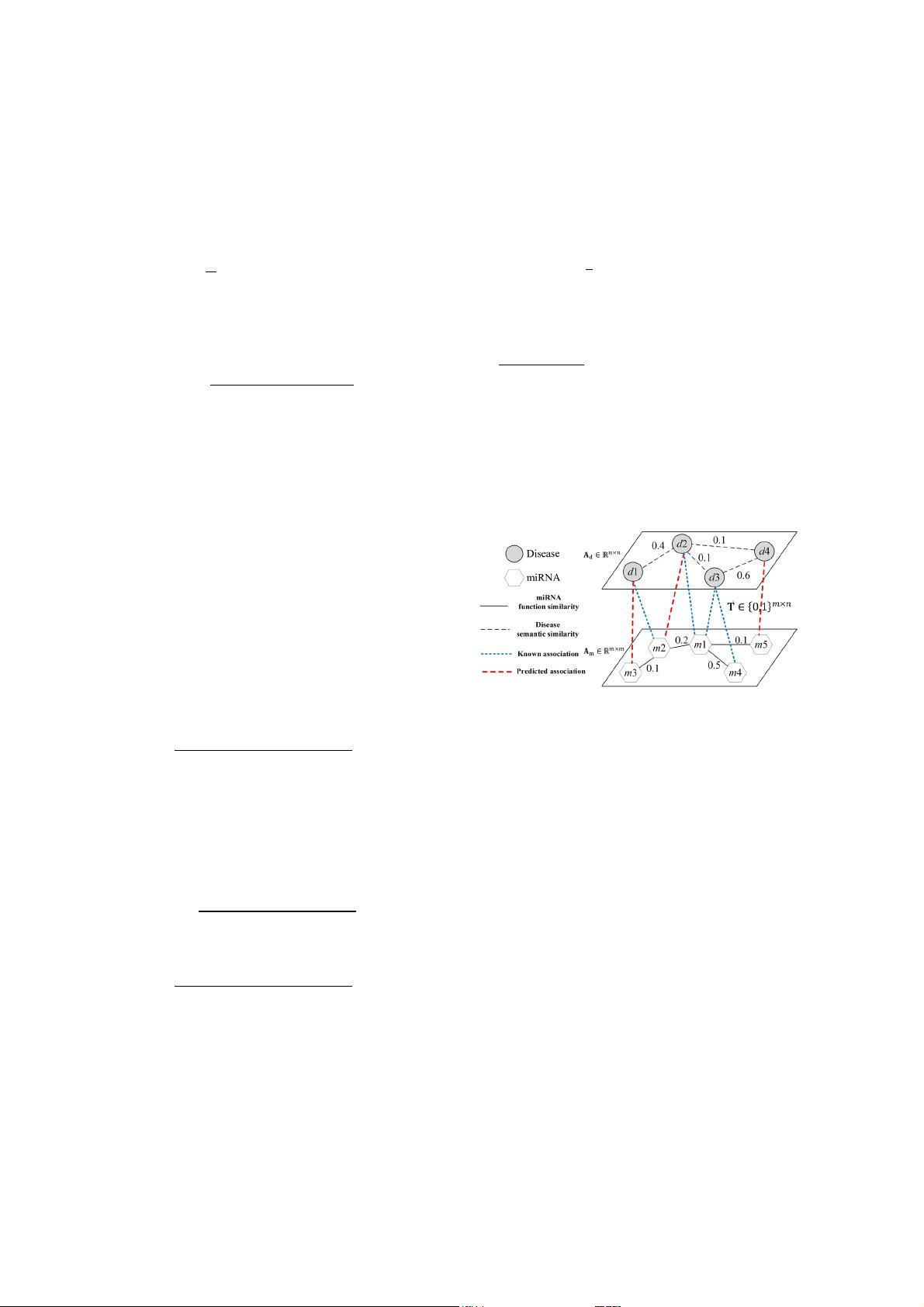
mation network as illustrated by the Fig.1. Note that both the miRNA
functional similarity network and the disease semantic similarity network
are edge-weighted graphs.
3 Methods
In this study, based on the miRNA-miRNA similarity network, the dis-
ease-disease similarity network, and the experimentally verified miRNA-
disease data, a novel NIMCGCN method was presented to effectively
solve the problem related to the prediction of miRNA-disease association.
3.1 Matrix Completion and Inductive Matrix Completion
A problem of miRNA-disease association prediction can be consid-
ered with 𝑚 miRNAs and 𝑛 diseases, and 𝑚×𝑛 experimentally verified
miRNA-disease association matrix 𝐓∈
0,1
×
. 𝐓
𝑖,𝑗
=1 if a miRNA
𝑖 is associated with a disease 𝑗. 𝐓
𝑖,𝑗
=0 if the association between 𝑖
and 𝑗 is unknown or unobserved. Without loss of generality, Ω and Ω
were
used to denote the set of observed and unobserved or unknown miRNA-
disease entries from the known association matrix 𝐓. The observation Ω
consisted only of positive associations, i.e., if ∀
𝑖,𝑗
∈Ω, 𝐓
𝑖,𝑗
=1. Ω
is the set of unknown or unobserved entries if ∀
𝑖,𝑗
∈Ω
, 𝐓
𝑖,𝑗
=0. A
sample of observed entries Ω from a true underlying matrix 𝐐 was consid-
ered. The objective was to estimate missing entries under some additional
assumptions on the structure of the association matrix 𝐓. The most com-
mon assumption is that 𝐐 is low-rank, i.e., 𝐐=𝐅𝐆
,where 𝐅∈ℝ
×
and
𝐆∈ℝ
×
are of rank 𝑘≪𝑚,𝑛. With these notations, the basic MDA can
be formulated as the following matrix completion problems:
Fig. 1. An Illustration of a miRNAs-diseases heterogeneous infor-
mation network
剩余13页未读,继续阅读
388 浏览量
603 浏览量
2021-03-24 上传
121 浏览量
128 浏览量
171 浏览量
117 浏览量
196 浏览量
185 浏览量

weixin_38725531
- 粉丝: 5
- 资源: 872
 我的内容管理
展开
我的内容管理
展开







最新资源
- Java职位面试之Java基础知识
- MPEG基础和协议分析指南
- RealTime OS Systems
- ATA-6 hard disk operation
- 微软软件测试面试考题
- c#数据结构 第一章概述ppt
- C++初学者的最佳资源PDF
- 长春理工大学应用光学课件.pdf
- MyEclipse+6+Java+开发中文教程_免费电子版.pdf
- 在VC中利用Kodak控件采集图像
- DB2数据库学习手册
- STL编程指南--详细的sgi参考手册
- 计算机网络统考串讲(习题部分)
- Oracle9i Database Administration Fundamentals I Ed 2.0.pdf
- unix C 字符串处理学习
- Oracle9i+数据库管理基础+IIVol.2.pdf
