RNA-Seq数据分析:揭示未注释的剪接 junctions 和转录多样性
下载需积分: 19 | PDF格式 | 1.73MB |
更新于2024-09-10
| 58 浏览量 | 举报
"这篇文章主要探讨了RNA数据分析,特别是RNA测序(RNA-Seq)在生物学研究中的应用及其带来的挑战。RNA-Seq技术可以提供全面的转录组图像,用于完整注释和量化任何样本中所有基因及其剪接异构体。尽管如此,充分利用这种技术需要复杂的计算方法。文章通过分析大量公开的RNA-seq数据集,揭示了人类基因剪接的多样性以及未被现有注释记录的剪接接头的广泛存在。"
在当前的生物学研究中,高通量RNA测序(RNA-Seq)已经成为探索复杂生物学问题的重要工具。与传统的分子生物学技术相比,RNA-Seq具有更高的灵敏度和分辨率,能够检测到转录组的细微变化,包括基因表达水平、转录剪接变异、非编码RNA以及罕见转录事件。RNA-Seq的工作流程通常包括样品准备、测序、数据生成、数据预处理、读取比对、定量分析和功能注释等步骤。
文章指出,尽管RNA-Seq技术潜力巨大,但随之而来的是对计算能力的需求增加。为了从海量的测序数据中提取有用信息,需要开发和应用各种生物信息学工具和算法,例如比对工具(如STAR、HISAT2)、转录本组装工具(如TransDecoder、Cufflinks)以及差异表达分析工具(如DESeq2、edgeR)。这些工具的使用旨在准确识别和量化基因表达,同时发现新的剪接变异和基因结构。
研究人员分析了来自Sequence Read Archive (SRA)的21,504个人类RNA-seq样本,将它们比对到人类基因组上,以评估与现有基因注释的符合程度。他们发现有56,861个剪接接头(约占18.6%)在至少1000个样本中未被现有GENCODE等基因注释所包含,并且这些未注释的剪接接头表达与特定组织类型相关。这表明,RNA-seq数据可以显著扩展我们对人类基因组剪接多样性的理解,揭示大量未被充分探索的遗传变异和转录事件。
此外,这项工作还强调了公共数据库如SRA在促进科研合作和数据共享方面的重要性。通过这样的大型数据分析,可以揭示在小规模研究中可能忽视的模式和趋势,进一步推动基因组学和转录组学的研究。
RNA数据分析,特别是RNA-Seq技术,对于揭示基因表达的复杂性和剪接的多样性具有重要意义。然而,它也带来了巨大的计算挑战,需要持续发展和优化计算方法来应对这些挑战。随着技术的不断进步和新工具的开发,我们可以期待更深入地理解基因功能和疾病机制,从而为精准医疗和药物研发提供新的见解。
Nellore et al. Genome Biology
(2016) 17:266
Page 3 of 14
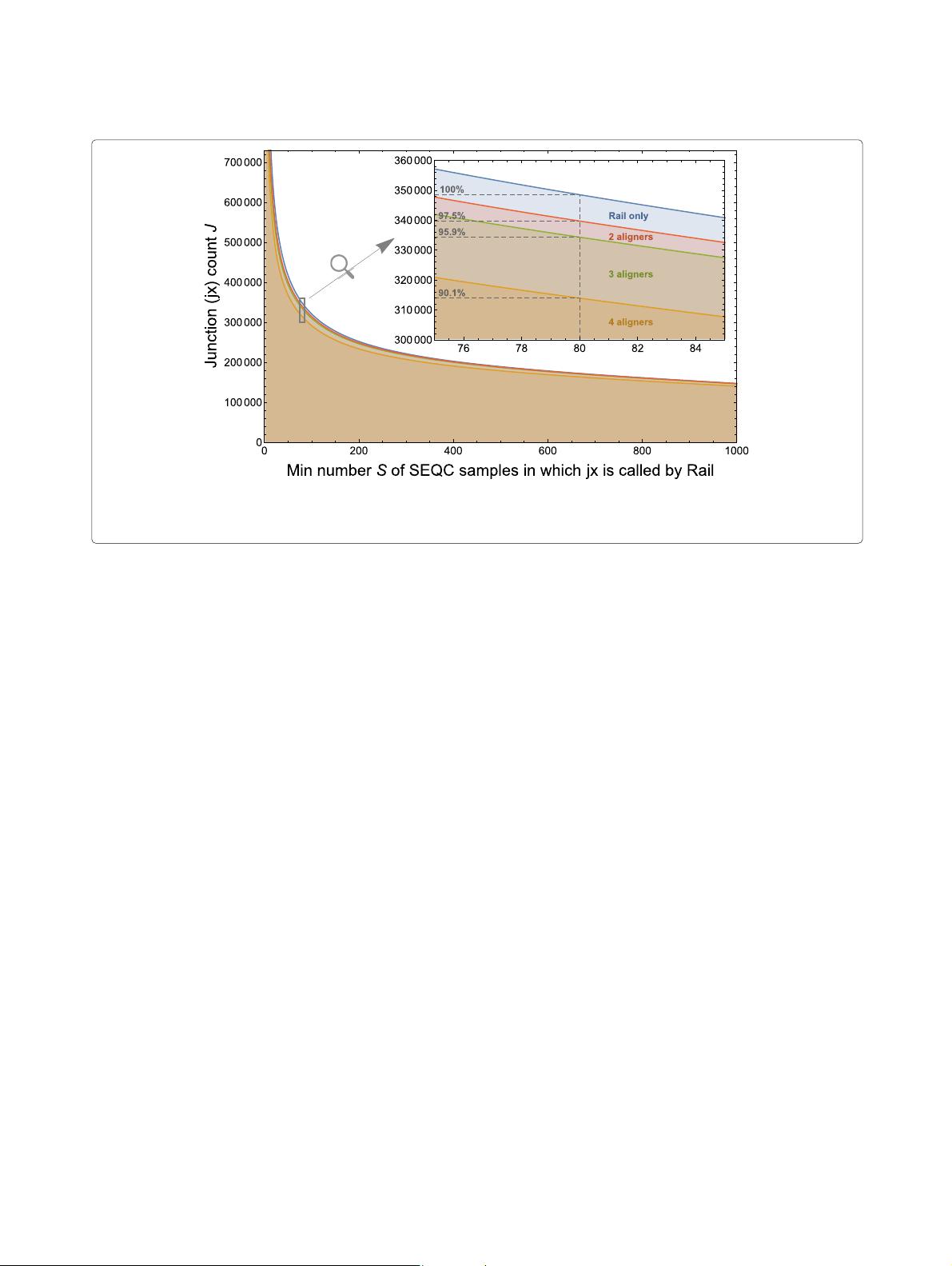
Fig. 1 Displayed is the number of exon-exon junctions J found by Rail-RNA and other alignment protocols in at least S of the 1720 brain and universal
human reference RNA-seq samples also studied by the SEQC/MACQ-III consortium [11] (i.e., SEQC). “2 aligners” (red), “3 aligners” (green), and “4
aligners” (orange) refer to junctions we found with Rail-RNA that were also found by, respectively, 1, 2, and 3 of the alignment protocols used by SEQC
of cutoffs. For each RNA-seq junction we considered, we
also evaluated whether it appeared in annotation. We con-
sidered the following levels of evidence: (1) fully annotated
junctions; (2) separately annotated junctions (typically
exon-skipping events), where both the donor and accep-
tor sites appear in one or more junctions from annotation,
but never for the same junction; (3) alternative donor and
acceptor sites, where only either the donor or the accep-
tor site appears in one or more junctions from annotation;
and (4) novel junctions, where neither donor nor acceptor
site is found in any annotated junction.
We observed that the RNA-seq junctions most widely
expressed across samples and experiments were well doc-
umented in annotation. For example, we observed that
junctions that appeared in at least 40% of human RNA-
seq samples on the SRA (S ≥ 8000) were also present in
previous annotation at least 99.8% of the time. However,
18.6% of junctions that appeared in 1000 or more sam-
ples did not appear in annotation (Fig. 2a). Many of these
unannotated junctions are partially annotated, but 3.5% of
junctions found in more than 1000 samples do not match
any splice site from an annotated junction.
We also took an investigator-focused view of the rela-
tionship between annotation and expression. Most inves-
tigators collect only a small number of samples for their
study. We restricted attention to samples where at least
100,000 RNA-seq junctions were found to rule out obvi-
ously small RNA-seq samples and samples that were
mislabeled as RNA-seq on the SRA. In each sample, we
counted the number of instances where a read maps
across a junction. (A read mapping across two junctions
thus contributes two instances.) The total number of such
“junction overlaps” across samples is a measure of the
total expression of junctions across the SRA. Most of
the reads that map to junctions map to annotated junc-
tions (Fig. 2b). In 10,090 of a total of 10,311 samples that
meet our criterion of 100,000 junctions observed, more
than 95% of junction overlaps correspond to annotated
junctions.
This represents only the bulk coverage of junctions.
We can also consider the number of junctions observed,
regardless of coverage. In 3389 out of 10,311 samples,
we observe that fewer than 80% of junctions appear in
annotation (Fig. 2c). So while the most highly covered
junctions are well annotated, there is a large number
of junctions that are well covered across multiple sam-
ples but may not appear in any given small subset of
samples.
To explore this idea further, we investigated the poten-
tial for single studies to be the sole contributors of individ-
ual unannotated junctions. In this event, the junction may
not have been called robustly across experimental proto-
cols. Here, we considered junctions that appeared in at
least P projects instead of samples. We again broke this
calculation down by the different potential levels of evi-
dence: whether the junction was entirely novel, had an
alternative donor or acceptor, an exon skip, or whether
it was fully annotated (Fig. 3). The story at the project
level mirrors the story at the sample level: 23.4% of junc-
tions found in more than 200 of the 929 projects are not
fully annotated. So unannotated junctions recur across
independent investigations.
剩余13页未读,继续阅读
相关推荐


202 浏览量

fengwanwan2017
- 粉丝: 0
 我的内容管理
展开
我的内容管理
展开







最新资源
- Ruby语言集成Mandrill API的gem开发
- 开源嵌入式qt软键盘SYSZUXpinyin可移植源代码
- Kinect2.0实现高清面部特征精确对齐技术
- React与GitHub Jobs API整合的就业搜索应用
- MATLAB傅里叶变换函数应用实例分析
- 探索鼠标悬停特效的实现与应用
- 工行捷德U盾64位驱动程序安装指南
- Apache与Tomcat整合集群配置教程
- 成为JavaScript英雄:掌握be-the-hero-master技巧
- 深入实践Java编程珠玑:第13章源代码解析
- Proficy Maintenance Gateway软件:实时维护策略助力业务变革
- HTML5图片上传与编辑控件的实现
- RTDS环境下电网STATCOM模型的应用与分析
- 掌握Matlab下偏微分方程的有限元方法解析
- Aop原理与示例程序解读
- projete大语言项目登陆页面设计与实现
