生物信息学进阶:NGS分析与资源指南
需积分: 49 140 浏览量
更新于2024-08-05
1
收藏 370KB DOCX 举报
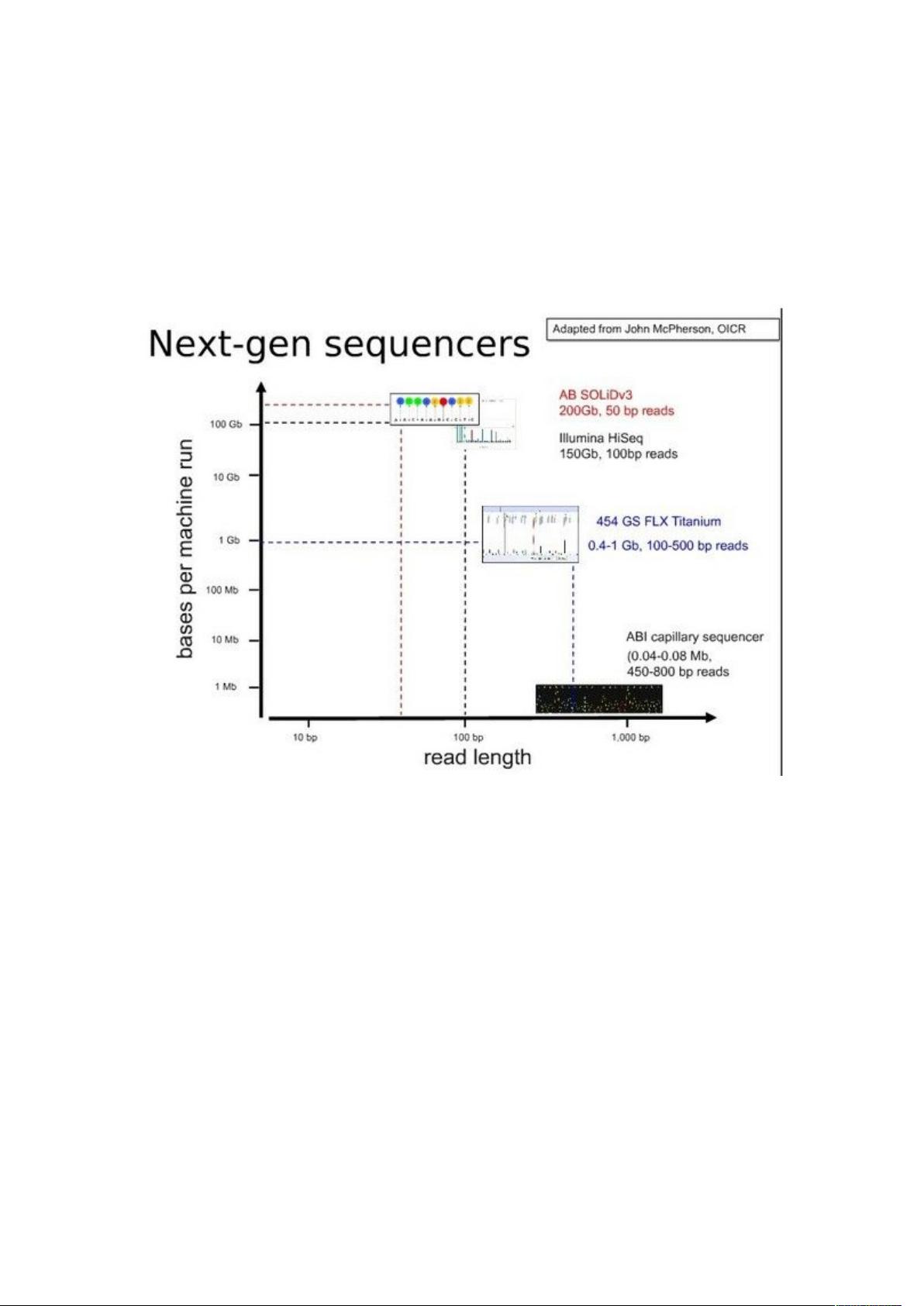
生物信息学进阶学习资源涵盖了二代测序数据预处理与分析的核心知识点,这对于深入理解生物信息学实践至关重要。二代测序技术(Next-generation sequencing, NGS)如 Illumina 和 PacBio 等,产生的大量数据需要经过一系列复杂步骤进行预处理和分析。
首先,数据质量控制是基础,FastQC 和 Fastx-toolkit 是常用的工具,用于检查读质量、 adapters 以及基本格式问题。拼接(Alignment)工具如 BWA、Bowtie、Tophat 和 SOAP2 负责将序列 reads 对齐到参考基因组,Mapper 如 Tophat 和 Cufflinks 则用于将 reads 映射到特定的基因区域。对于基因表达定量,Cufflinks 和 AvadisNGS 可以进行定量分析,而 Genome Analysis Toolkit (GATK) 则提供了质量改进的功能,包括 SNP 分析。
SNP(Single Nucleotide Polymorphism)检测有多种工具可用,如 Unified Genotyper、GATK 的 GLF Multiple、SAMtools 和 AvadisNGS。CNV(Copy Number Variation)分析则可以通过 CNVnator 进行,而 Indel(插入/缺失)检测有 Pindel、Dindel、Unified Genotyper 和 AvadisNGS。此外,如需将 reads 映射到具体的基因,Cufflinks 和 Rsamtools 也可派上用场,GenomicFeatures 则提供更细致的基因组功能。
数据格式方面,FASTQ 用于存储原始序列数据,SAM 是通用的核苷酸比对格式,而 BAM 是其二进制形式,方便存储和索引。VCF(Variant Call Format)是遗传变异报告的标准格式。RNA-seq 数据因其高度可变性和基因数量庞大,使得分析相对复杂,同时样本量小和巨大变异也增加了挑战。
关于SNP的鉴定,虽然DNA-seq和RNA-seq都能检测,但RNA-seq额外要考虑可变剪接带来的SNP,这可能会影响最终的生物学解释。利用 Barcode 序列对 fastq 文件进行拆分,是处理混杂样本的重要步骤,如 seqtk_demultiplex 和 fastq-multx 是两个常用的工具。
在肿瘤标记物预测方面,当前存在一些局限。首先,大部分研究发现的肿瘤标志物具有一定的关联性但非特异性,因此在选择时需要考虑排除其他疾病的影响,以提高筛查的准确性。其次,很多研究未能充分考虑肿瘤标志物与恶性程度(如分期、分型和生存率)之间的关系,这是未来研究的一个重要发展方向。
这个资源文档为生物信息学从业者提供了全面的二代测序数据分析指南,包括工具选择、数据处理流程和实际应用中的关键注意事项,对于进一步提升生物信息学技能具有很高的价值。
二代测序数据预处理与分析
Next generation sequencing (NGS)
常使用的工具列表
质量控制 Quality Control:FastQC、Fastx-toolkit
拼接 Aligner:BWA,Bowtie, Tophat, SOAP2
Mapper:Tophat, Cu!inks
基因定量 Gene Quanti#cation: Cu!inks, Avadis NGS
质量改进 Quality improvement:*Genome Analysis Toolkit(GATK)
SNP: Uni#ed Genotyper,Glfmultiple, SAMtools, Avadis NGS
CNV: CNVnator
Indel: Pindel, Dindel, Uni#ed Genotyper, Avadis NGS
Mapping to a gene: Cu!inks, Rsamtools,*Genomic Features
下载后可阅读完整内容,剩余6页未读,立即下载

qi201910
- 粉丝: 0
- 资源: 1
 我的内容管理
展开
我的内容管理
展开







最新资源
- model_MEPERS
- Contacts_App
- java版商城源码-learnUrl:学习网址
- paizhao.zip
- 新星
- ACs---Engenharia:为需求工程主题的AC1创建的存储库
- tmux-power:mu Tmux电力线主题
- Flutter_frist_demo:颤振学习演示
- java版商城源码-mall:购物中心
- u5_final
- 华为模拟器企业网设计.zip
- python-random-integer-project
- aqi-tool:空气质量指数(AQI)计算器
- java版商城源码-MachiKoroDigitization:MachiKoro游戏由3人组成
- c04-ch5-exercices-leandregrimmel:c04-ch5-exercices-leandregrimmel由GitHub Classroom创建
- Monique-Nilles
