VASP程序使用详解:从基础到高级
需积分: 9 122 浏览量
更新于2024-07-30
收藏 2.72MB PDF 举报
"VASP使用指南,由复旦大学候住峰提供,涵盖了程序原理、输入输出文件、选择POTCAR文件、LDA与GGA、基本任务等内容,旨在指导用户进行第一原理电子结构计算。"
VASP(Vienna Ab initio Simulation Package)是一款广泛应用于材料科学领域的第一原理量子力学计算软件,它基于密度泛函理论(DFT),用于模拟固体的电子结构、能量、结构优化等。在使用VASP进行计算前,理解其工作原理和输入输出文件至关重要。
程序原理:
VASP利用平面波基组和赝势方法处理电子结构问题。平面波基组用于描述电子波函数,而赝势则模拟了原子核和内层电子对价电子的影响。VASP采用 projector-augmented wave (PAW) 方法,该方法在保持计算精度的同时减少了计算复杂性。
输入文件:
1. POTCAR:包含了原子的赝势信息,如元素类型、核心状态、交换关联泛函等。例如,"LEXCH=CA"表示使用CA交换关联,"GGA=-1.4125-1.4408.0293-.9884eV"定义了GGA参数。
2. KPOINTS:控制布里渊区采样,如采用Monkhorst-Pack网格,"111111"定义了k点网格大小。
3. POSCAR:存储晶体结构数据,包括晶格常数、原子种类、原子数量以及原子在基矢坐标系下的位置。
4. INCAR:设定计算的控制参数,如"System=diamondSi"定义系统为金刚石硅,"ENCUT=150.0"设定了能量截断值,"NELM=200"是迭代次数,"IBRION=2"选择结构优化算法。
应用:
1. 计算电子态密度和能带:这些信息可以揭示材料的电子性质,如导电性、磁性和光学性质。
2. 优化晶体参数:通过最小化能量找到稳定结构,例如,"ISIF=2"表示同时优化原子位置和体积。
3. 内部自由度弛豫:包括原子坐标和晶格参数的优化,"ISIF=2"配合"IBRION=2"实现这一目标。
4. 结构弛豫:自动调整原子位置以达到能量最小化,"ISYM=1"开启对称性考虑,"LWAVE=F"和"LCHARG=F"关闭波函数和电荷密度的输出。
输出文件主要包括OUTCAR、CONTCAR(优化后的结构文件)和OSZICAR(能量和其他关键参数的变化历史)。通过分析这些输出文件,用户可以获取到计算结果和系统行为的详细信息。
在使用VASP时,正确选择POTCAR文件是至关重要的。例如,LDA(Local Density Approximation)和GGA(Generalized Gradient Approximation)是两种常见的交换关联泛函,其中PAW_LDA和PAW_GGA分别对应LDA和GGA的PAW赝势。在实际计算中,根据研究对象的特性选择合适的POTCAR和交换关联函数能够提高计算的准确性。
理解并熟练掌握VASP的输入输出文件和相关参数设置,是成功进行第一原理计算的关键。通过候住峰的"VASP使用指南",用户将能够更深入地理解和应用这个强大的计算工具。
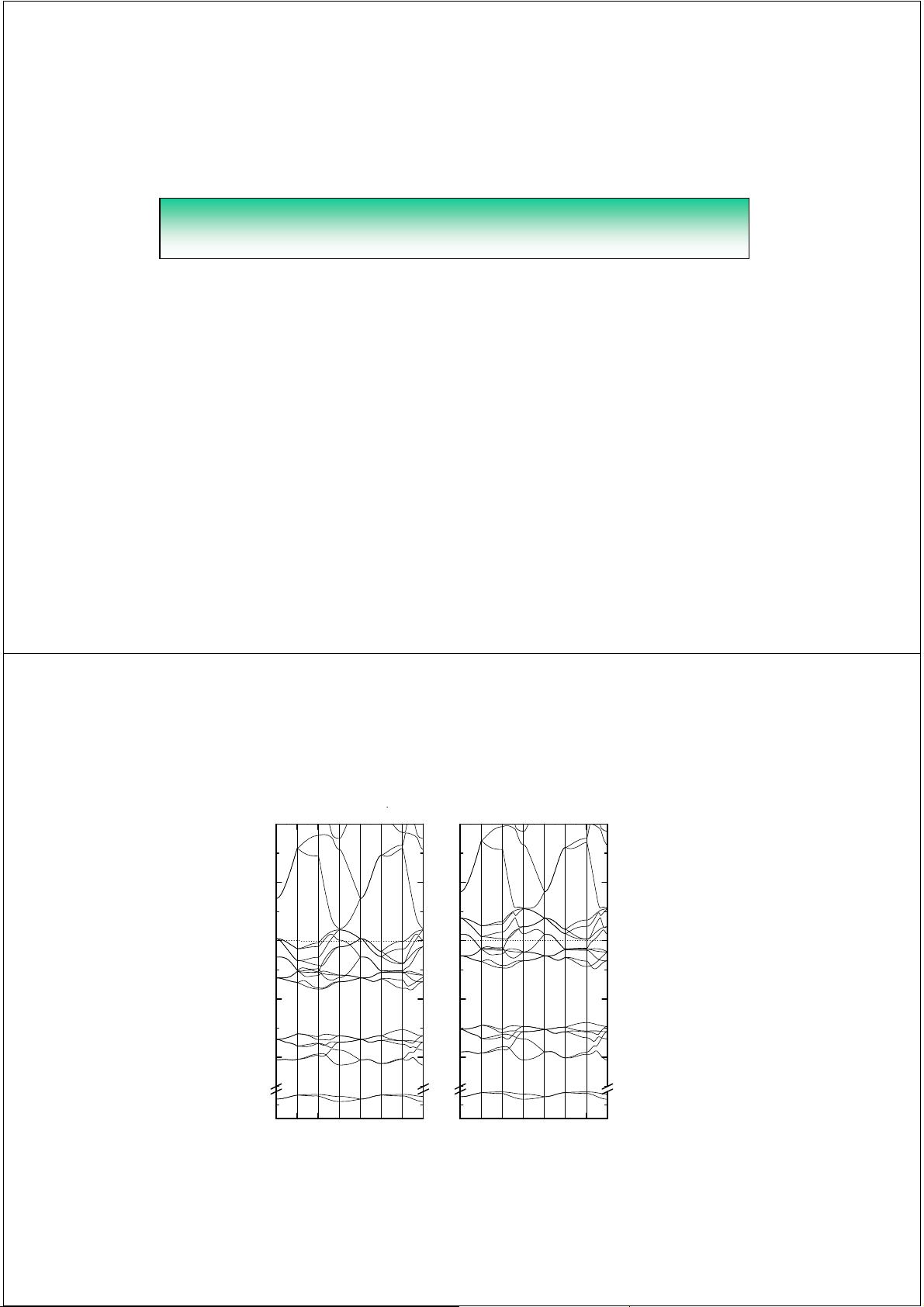
(2). 优化晶格参数,求出能量最低所对应的晶格参数
wurtzite晶体含有两个内部自由度, 晶格参数优化过程要比立方
结构费时
CoO: a=2.98, c/a=1.735, u=0.367
-24
-8
-4
0
4
8
-24
-8
-4
0
4
8
A L M Γ A H K Γ
A L M Γ A H K Γ
E (eV)



剩余144页未读,继续阅读
2012-11-22 上传
2012-11-09 上传
2011-03-10 上传
2022-08-04 上传
点击了解资源详情
点击了解资源详情
点击了解资源详情

wang_f_lu
- 粉丝: 0
- 资源: 7
 我的内容管理
展开
我的内容管理
展开







最新资源
- C语言初级学习100例 pdf文件
- Linux内核完全注释(内核版本0.11)
- 银川技能大赛试题园区网
- display标签使用
- Apress Foundation Expression Blend 2 Building Applications in WPF and Silverlight 2008
- IC封装大全IC封装大全
- C#.net打包时自定义应用程序的快捷方式与卸载
- WinCC手册1.pdf
- 信息隐藏检测lsb matching
- CCNA笔记精简整理版
- Berkeley DB彻底了解(存取方式、各种API、例子)
- java实现的b/s权限管理系统----<下载不要分,回帖加1分,欢迎下载,童叟无欺>
- 悟透JavaScript
- 在Visual C#中使用XML指南之读取XML
- 解析.Net框架下的XML编程技术
- HTML超文本标记语言教程
