《从基础到高级:Ab Initio分子动力学理论与实践指南》
"《第一原理分子动力学:基本理论与高级方法》是由Dominik Marx和Jürg Hutter共同编著的一本专著,由剑桥大学出版社出版。这本书是分子动力学与电子结构理论结合领域的一个里程碑,它革新了复杂分子系统和过程(如化学反应)的计算机模拟。书中对从头算(ab initio)分子动力学这一前沿技术进行了全面而深入的探讨。
本书旨在为研究生和研究人员提供一个系统的介绍,涵盖了从基础理论到高级方法的广泛范围。作者精心设计了一系列循序渐进的讲解,让读者能够透彻理解并评估常用的ab initio分子动力学技术的优势和局限性。通过这种方法,研究者可以避免陷入目前科研文献中常见的误解,从而更准确地应用这些技术。
特别值得一提的是,书中对广泛应用的Car-Parrinello方法进行了详尽剖析,澄清了该方法的一些常见误区。这不仅对于那些初次接触该领域的新人来说是宝贵的教育资源,也对现有程序开发者的优化工作提供了实用的指导。书中还包含了一些典型平面波电子结构代码的伪代码和程序布局,帮助读者熟悉常用软件包的工作原理,并激发开发者对代码进行改进和扩展的能力。
《第一原理分子动力学:基本理论与高级方法》是一本不可或缺的参考书籍,不仅适用于深化理论研究,而且对于实际操作中的模拟和计算实践具有重要的指导意义,对于推动整个领域的发展起到了关键作用。"
4 Setting the stage: why ab initio molecular dynamics?
1970 1980 1990 2000 2010
Year n
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
11000
12000
Number N
CP PRL 1985
AIMD
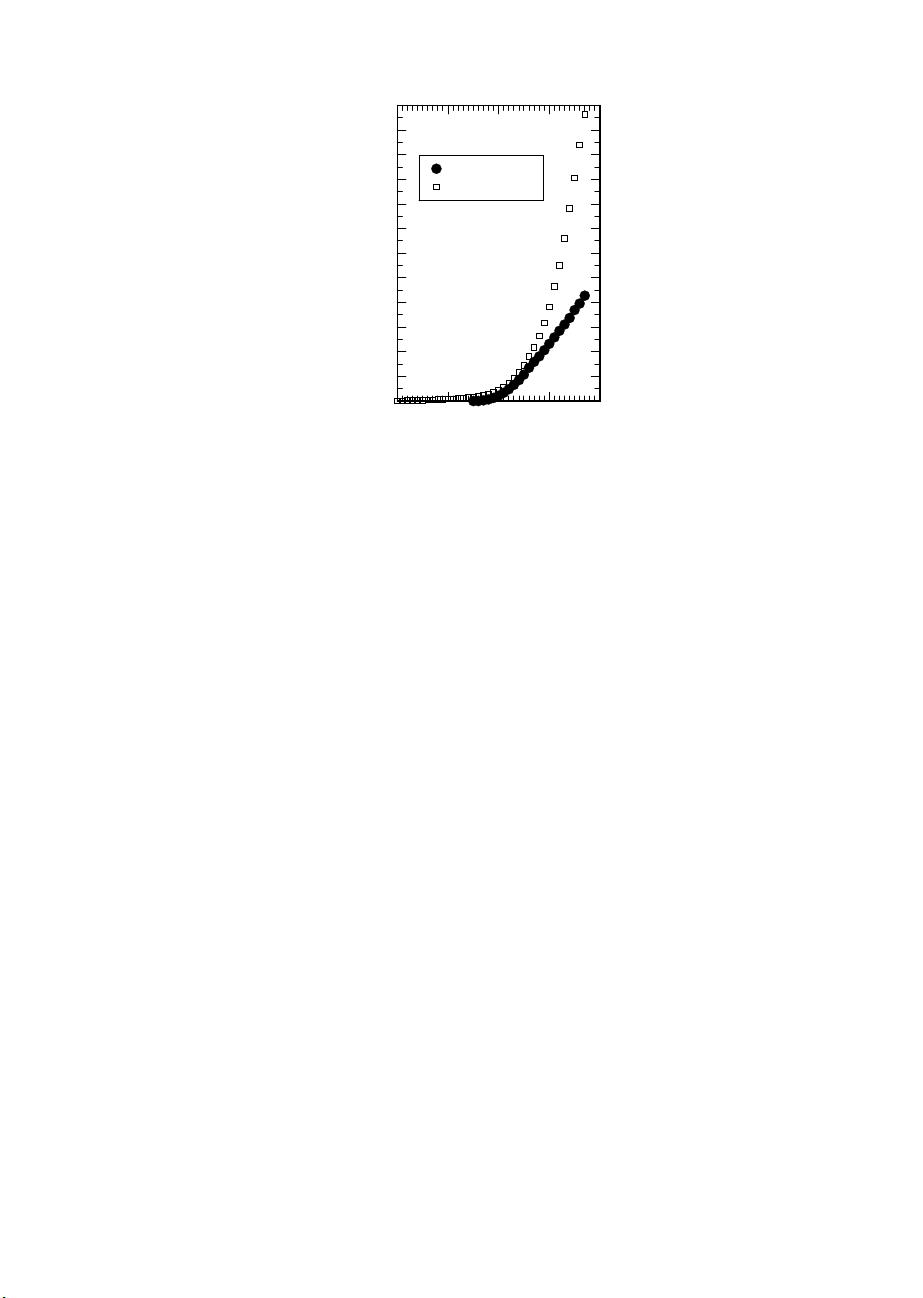
Fig. 1.2. Publication and citation analysis up to the year 2007. Squares: number of
publications N which appeared up to the year n containing the keyword “ab initio
molecular dynamics” (or synonyms such as “first principles MD”, “Car–Parrinello
simulations” etc.) in title, abstract or keyword list. Circles: number of publications
N which appeared up to the year n citing the 1985 paper by Car and Parrinello [222]
(including misspellings of the bibliographic reference). Self-citations and self-papers
are excluded, i.e. citations of Ref. [222] in their own papers and papers coauthored
by R. Car and/or M. Parrinello are not considered in the respective statistics;
note that this, together with the correction for misspellings, is probably the main
reason for a slightly different citation number up to the year 2002 as given here
compared to that (2819 citations) reported in Ref. [1216]. The analysis is based
on Thomson/ISI Web of Science (WoS), literature file CAPLUS of the Chemical
Abstracts Service (CAS), and INSPEC file (Physics Abstracts) as accessible under
the database provider STN International. Earlier reports of these statistics [933,
934, 943] are updated as of March 13, 2008; the authors are most grateful to
Dr. Werner Marx (Information Service for the Institutes of the Chemical Physical
Technical Section of the Max Planck Society) for carrying out these analyses.
(Car–Parrinello) and other numerical simulations”) introduced in 1996 into
the Physics and Astronomy Classification Scheme [1093].
Despite its obvious advantages, it is evident that a price has to be payed
for putting molecular dynamics onto an ab initio foundation: the corre-
lation lengths and relaxation times that are accessible are much smaller
than what is affordable in the framework of standard molecular dynamics.
More recently, this discrepancy was counterbalanced by the ever-increasing
power of available computing resources, in particular massively parallel plat-
forms [661, 662], which shifted many problems in the physical sciences right
into the realm of ab initio molecular dynamics. Another appealing feature
of standard molecular dynamics is less evident, namely the experimental




剩余578页未读,继续阅读
1206 浏览量
266 浏览量
148 浏览量
392 浏览量
2021-05-26 上传
2021-05-26 上传
点击了解资源详情
154 浏览量

hchfoxforum
- 粉丝: 0
 我的内容管理
展开
我的内容管理
展开







最新资源
- 网页自动刷新工具 v1.1 - 自定义时间间隔与关机
- pt-1.4协程源码深度解析
- EP4CE6E22C8芯片三相正弦波发生器设计与实现
- 高效处理超大XML文件的查看工具介绍
- 64K极限挑战:国际程序设计大赛优秀3D作品展
- ENVI软件全面应用教程指南
- 学生档案管理系统设计与开发
- 网络伪书:社区驱动的在线音乐制图平台
- Lettuce 5.0.3中文API文档完整包下载指南
- 雅虎通Yahoo! Messenger v0.8.115即时聊天功能详解
- 将Android手机转变为IP监控摄像机
- PLSQL入门教程:变量声明与程序交互
- 掌握.NET三层架构:实例学习与源码解析
- WPF中Devexpress GridControl分组功能实例分析
- H3Viewer: VS2010专用高效帮助文档查看工具
- STM32CubeMX LED与按键初始化及外部中断处理教程
