Gaussian03技术详解:分子坐标与计算化学应用
需积分: 10 60 浏览量
更新于2024-08-02
收藏 544KB PPT 举报
"Gaussian——PPT——2:讲解了Gaussian软件的主要操作及其在计算有机化学中的应用,包括分子坐标、基组选择、错误处理等,并涉及计算化学中的自旋多重度概念及其判断方法。"
Gaussian是一款广泛应用在计算化学领域的软件,它能进行各种分子动力学模拟、量子力学计算以及反应路径分析等任务。Gaussian03是其特定的版本,提供了丰富的功能和优化的计算方法。
在Gaussian03中,输入文件的设置至关重要。例如,`%chk=XXX`用于指定检查文件名,保存计算过程中的中间数据;`%MEM=XXXXMBorGBorMW`定义内存分配;`%RWF`则用于指定波函数文件的位置和大小。此外,还有 `%SCR=XXX.scr` 指定临时文件,`%D2E=xxx.d2e` 用于二阶微扰能量文件,`%INT=xxx.int` 用于积分文件,`%KjobL301(n)` 设置作业参数,`%Nosave` 防止保存输出,`%nproc`, `%nproclinda`, `%nprocshared` 分别设置并行计算的核心数。
分子描述部分,我们关注的是分子的电荷和自旋多重度。电荷指分子整体的净电荷,而自旋多重度是由分子的电子自旋状态决定的,与分子的稳定性密切相关。自旋多重度(2S+1)是基于自旋量子数S计算得出的,S为电子总自旋在Z轴上的分量。对于闭壳层体系,自旋多重度为1;单电子体系,自旋多重度为2。成对的电子对自旋多重度没有贡献。判断自旋多重度的例子包括一、二周期原子,过渡金属如Mn、Fe等,需要考虑其电子排布和自旋状态。
在实际应用中,分子结构的输入可以通过多种方式实现,如手动编写内坐标、使用ChemDraw、GaussView或Molden等分子绘图软件生成,或者从实验数据如晶体结构数据中导入。
Gaussian软件在计算化学中扮演着重要角色,不仅处理基础的计算任务,还能深入分析分子的性质,如自旋多重度,这对于理解分子的电子结构和反应机理至关重要。掌握Gaussian的正确使用和参数设定,能够极大地提升化学研究的效率和精确度。
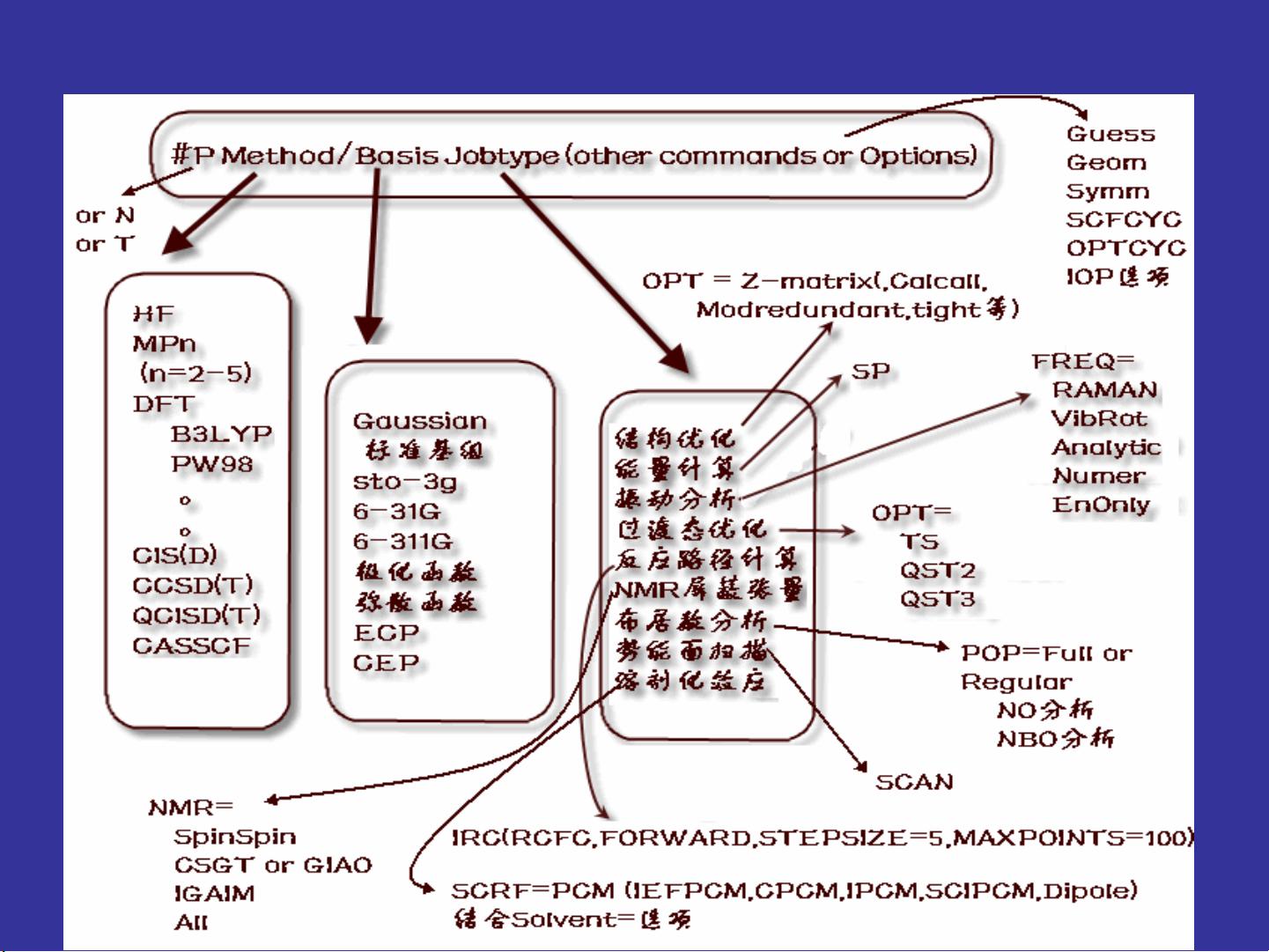
基本输入命令 :Job_Command Line

剩余43页未读,继续阅读
2021-01-11 上传
2021-10-03 上传
2023-05-27 上传
2023-05-27 上传
2023-07-28 上传
2023-03-29 上传
2023-05-27 上传
2024-09-12 上传

hchuanqi
- 粉丝: 1
- 资源: 2
 我的内容管理
展开
我的内容管理
展开







最新资源
- 掌握Jive for Android SDK:示例应用的使用指南
- Python中的贝叶斯建模与概率编程指南
- 自动化NBA球员统计分析与电子邮件报告工具
- 下载安卓购物经理带源代码完整项目
- 图片压缩包中的内容解密
- C++基础教程视频-数据类型与运算符详解
- 探索Java中的曼德布罗图形绘制
- VTK9.3.0 64位SDK包发布,图像处理开发利器
- 自导向运载平台的行业设计方案解读
- 自定义 Datadog 代理检查:Python 实现与应用
- 基于Python实现的商品推荐系统源码与项目说明
- PMing繁体版字体下载,设计师必备素材
- 软件工程餐厅项目存储库:Java语言实践
- 康佳LED55R6000U电视机固件升级指南
- Sublime Text状态栏插件:ShowOpenFiles功能详解
- 一站式部署thinksns社交系统,小白轻松上手
