2011年Gromacs分子模拟讲座:原理与应用详解
需积分: 9 27 浏览量
更新于2024-07-15
收藏 2.38MB PDF 举报
Gromacs-lec-2011-11-session1.pdf是一份关于Gromacs程序的详细介绍文档,由清华大学机械系的李启楷教授在2011年的高性能计算培训中分享。Gromacs是一款专用于分子生物学和生物化学领域的分子动力学模拟工具,其核心优势包括:
1. **基础原理与应用**:该讲座首先概述了Gromacs的基本原理,它是如何将物理、化学和生物学理论链接起来,通过模拟难以在实验中观察的现象,如蛋白质折叠和纳米尺度过程,来理解这些复杂系统的行为。
2. **软件特性**:
- **编程语言与并行处理**:Gromacs主要用C语言编写,同时结合MPI库和线程进行并行化处理,使其能够高效地运行在分布式内存集群和共享内存计算机上。
- **内置功能**:提供多种内置的力场和工具,支持各种类型的分子模拟研究。
- **免费下载**:最新版本(4.5.5)是开源的,可以从gromacs.org网站免费获取。
3. **模拟目的**:MD(分子动力学)模拟之所以重要,是因为它基于牛顿运动定律,通过微小时间步长的积分来计算粒子的轨迹,模拟系统随时间的动态变化。这使得研究人员能够计算关键的热力学量,如自由能和结合能,从而深入了解分子间相互作用和系统行为。
4. **模拟方法**:利用牛顿方程,特别是动量定理,通过计算每个粒子受到的力(F),根据牛顿第二定律(F = ma),确定加速度,然后通过微分来估计位置变化。这个过程被重复执行,生成系统的动力学轨迹,从而揭示分子层面的动态行为。
文件内容涵盖了Gromacs在分子模拟中的基本概念、技术细节以及其在科学研究中的实际应用,对想要学习或使用Gromacs进行科研的人员来说,这是一个宝贵的资源。通过这份讲座,参与者可以掌握如何设置和运行Gromacs模拟,以及如何解读和分析模拟结果,从而推动生物学和化学领域的理论进步。
16
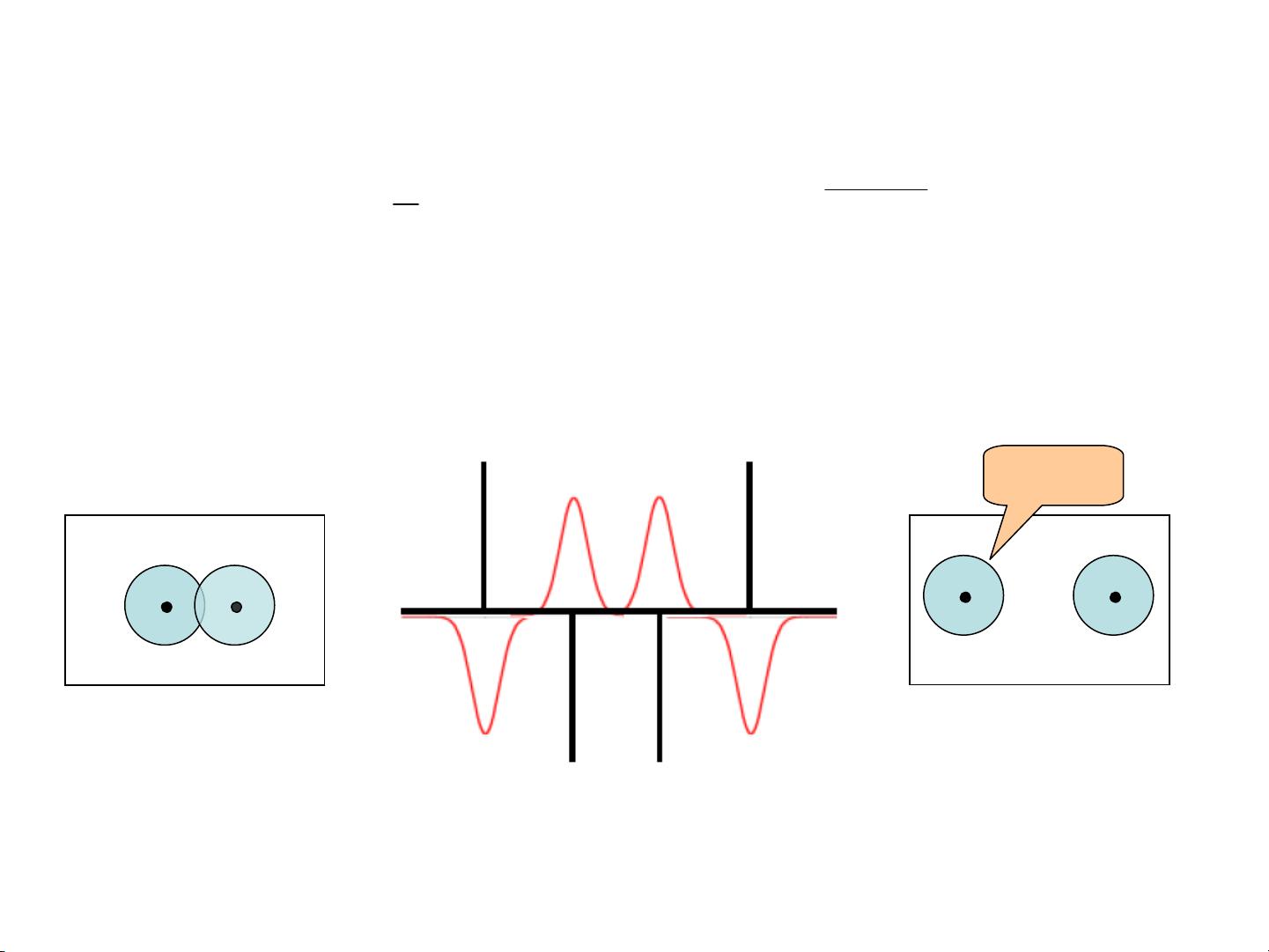
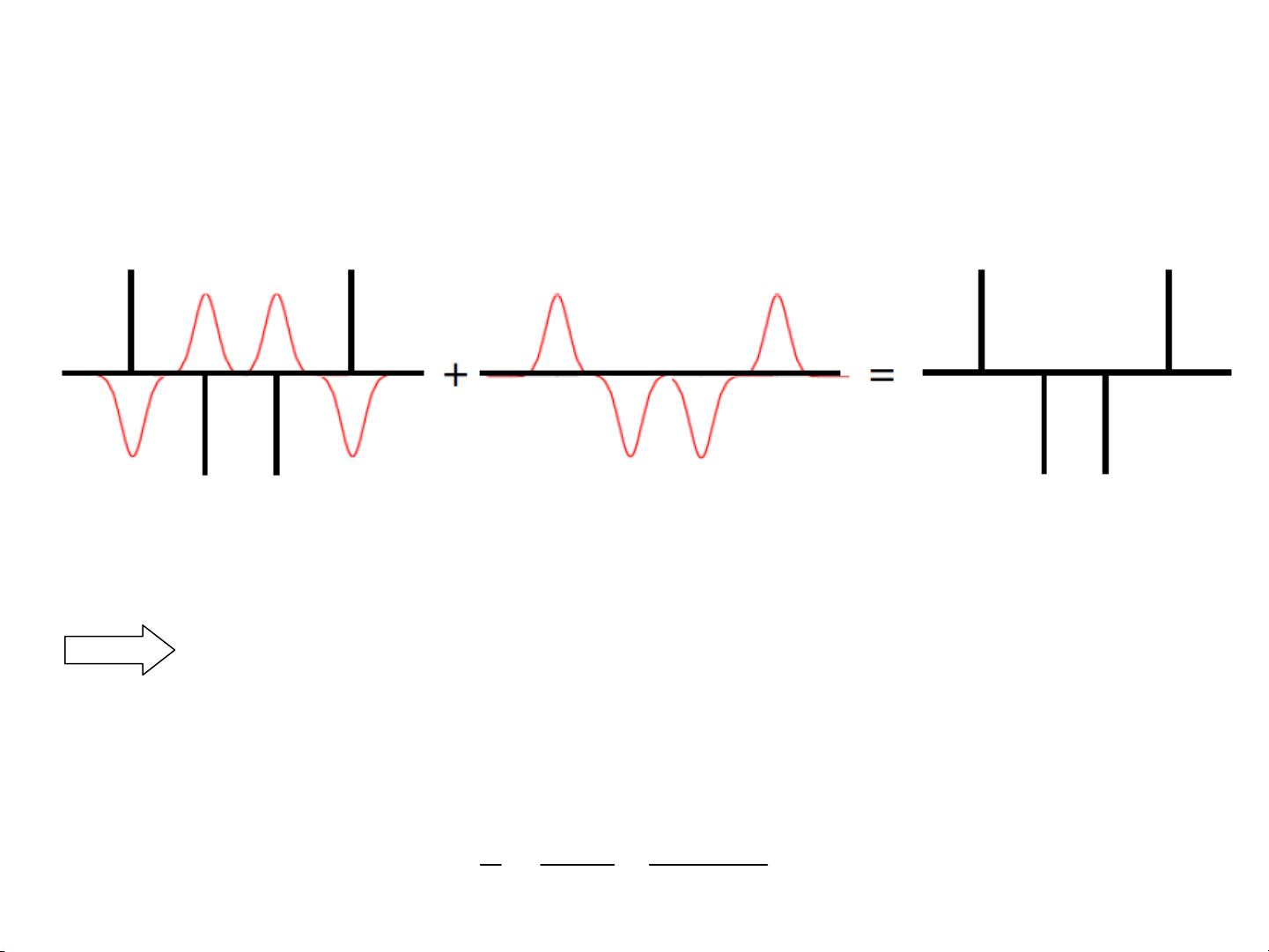
Nonbonded Interactions
Modified non-bonded interactions
In the GROMACS force field, the non-bonded potentials can be modified by a shift
function. The purpose of this is to replace the truncated forces by forces that are continuous
and have continuous derivatives
at the cutoff radius. With such forces the time-step
integration produces much smaller errors and there are no such complications as creating
charges from
dipoles
by the truncation procedure.
In fact, by using shifted forces there is no need for charge groups
in the construction of
neighbor lists.
However, the shift function produces a considerable modification
of the Coulomb
potential.
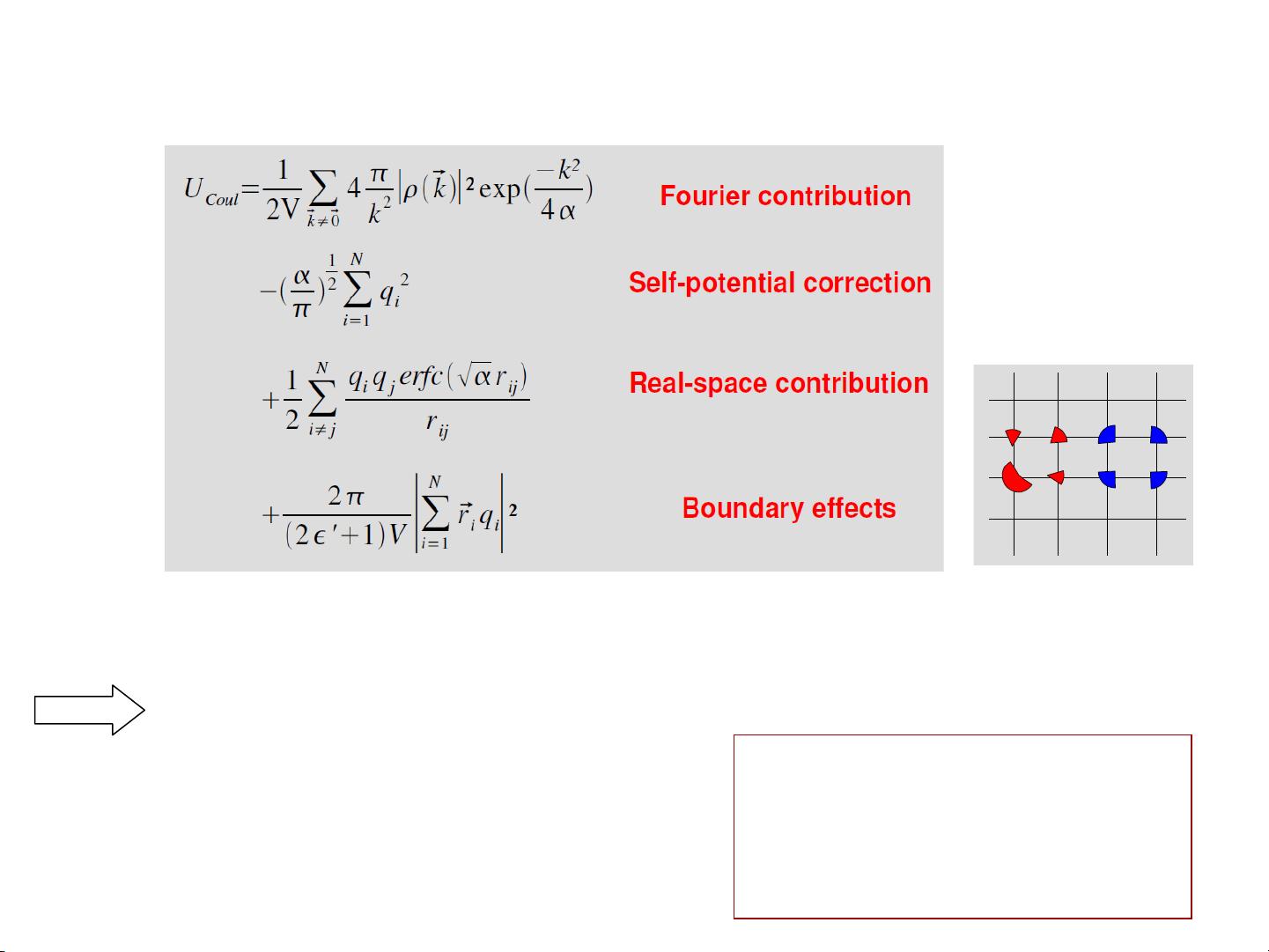
Unless the ‘missing’
long-range potential is properly calculated and added
(through the use of PPPM, Ewald, or PME), the effect of such modifications must be
carefully evaluated.
The modification of the Lennard-Jones dispersion and repulsion is only minor, but it does
remove the noise caused by cutoff effects.
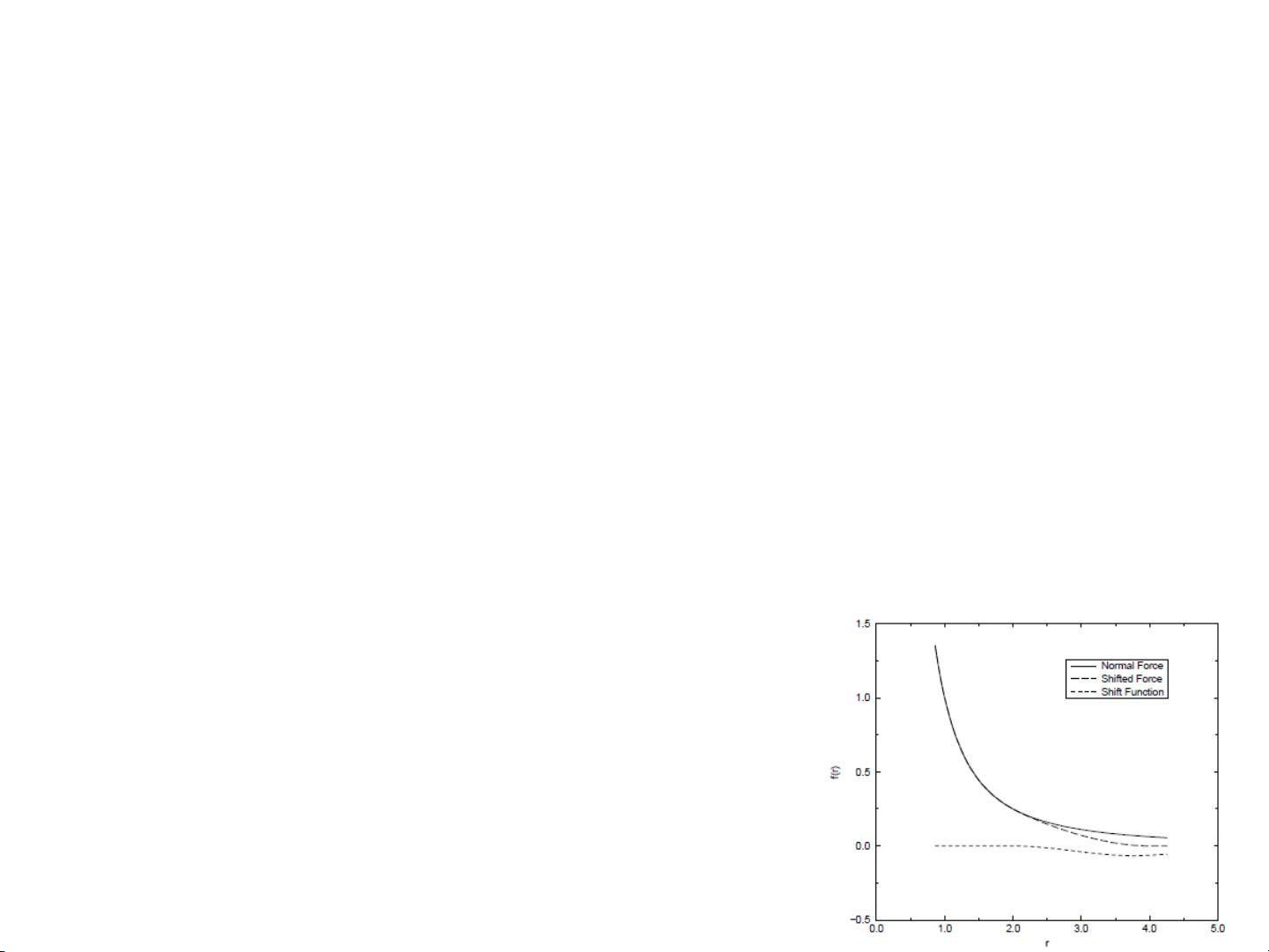
Figure 4.4: The Coulomb Force, Shifted Force and Shift
Function S(r), using r
1
= 2 and r
c
= 4.
剩余96页未读,继续阅读
2020-07-14 上传
2021-04-22 上传
2021-05-03 上传
2024-03-28 上传
2024-03-28 上传
2020-07-14 上传
2024-03-28 上传
2020-12-28 上传
2021-04-26 上传

D2472260125
- 粉丝: 1
- 资源: 35
 我的内容管理
展开
我的内容管理
展开







最新资源
- MATLAB新功能:Multi-frame ViewRGB制作彩色图阴影
- XKCD Substitutions 3-crx插件:创新的网页文字替换工具
- Python实现8位等离子效果开源项目plasma.py解读
- 维护商店移动应用:基于PhoneGap的移动API应用
- Laravel-Admin的Redis Manager扩展使用教程
- Jekyll代理主题使用指南及文件结构解析
- cPanel中PHP多版本插件的安装与配置指南
- 深入探讨React和Typescript在Alias kopio游戏中的应用
- node.js OSC服务器实现:Gibber消息转换技术解析
- 体验最新升级版的mdbootstrap pro 6.1.0组件库
- 超市盘点过机系统实现与delphi应用
- Boogle: 探索 Python 编程的 Boggle 仿制品
- C++实现的Physics2D简易2D物理模拟
- 傅里叶级数在分数阶微分积分计算中的应用与实现
- Windows Phone与PhoneGap应用隔离存储文件访问方法
- iso8601-interval-recurrence:掌握ISO8601日期范围与重复间隔检查
