LAMMPS模拟指南:从入门到精通
下载需积分: 21 | PDF格式 | 4.59MB |
更新于2024-07-09
| 41 浏览量 | 举报
"60分钟实现LAMMPS素人逆袭.pdf"
LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)是一款广泛用于分子动力学模拟的开源软件,尤其适合大规模并行计算。该软件由美国劳伦斯伯克利国家实验室开发,可用于模拟各种物质系统,包括固体、液体、表面、生物大分子等,涵盖了材料科学、化学、物理等多个领域。
LAMMPS模拟原理基于牛顿第二定律,即物体的运动状态由作用在其上的力决定。在分子动力学模拟中,LAMMPS通过数值方法求解粒子系统的牛顿方程,追踪每一个粒子的位置和速度随时间的变化。它采用了多种时间步进算法,如Verlet算法,以确保模拟的稳定性和精度。
LAMMPS的模拟用途广泛,可以用来研究物质的形变、相变、结晶过程、扩散、反应动力学等现象。通过模拟,科研人员能够预测材料的性质,优化材料设计,甚至在微观层面上理解复杂的物理和化学过程。
LAMMPS的使用流程主要包括以下几个步骤:
1. **建模**:根据研究目标构建原子或分子模型,这可能涉及到选择合适的原子类型、确定初始构型、设置边界条件等。
2. **势能参数获取**:选择合适的势函数(如Lennard-Jones、EAM、REAXFF等),并获取对应的势参数。这些参数通常可以从文献中获取或者使用预训练的参数库。
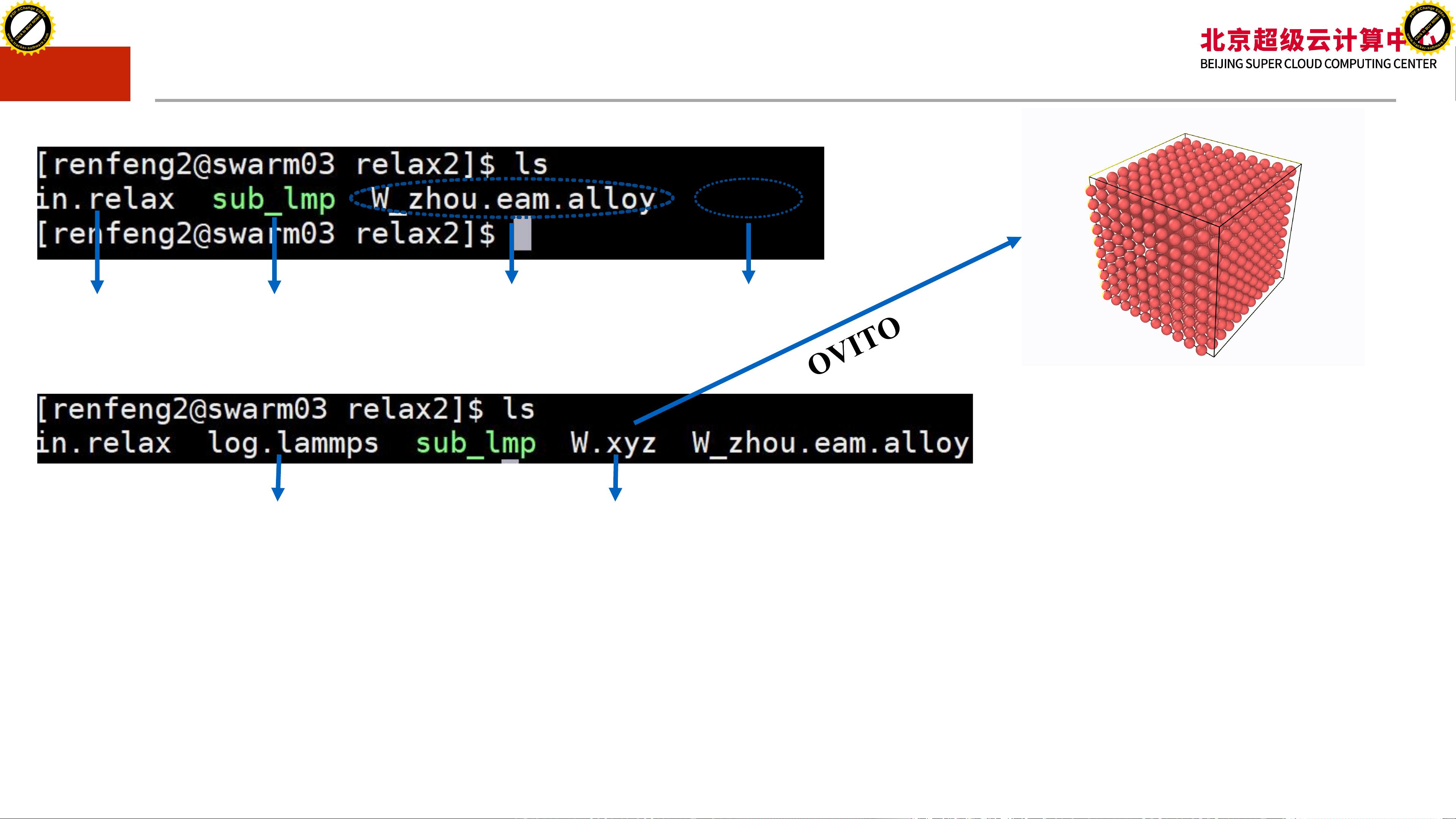
3. **编写in文件**:LAMMPS的输入文件(in文件)是模拟的核心,包含了所有模拟设置,如时间步长、模拟时间、力场信息、热力学输出等。
4. **运行LAMMPS**:执行LAMMPS程序,进行模拟计算。
5. **后处理分析**:对输出数据进行分析,如能量、压力、结构因子等,以获得科学研究所需的结果。
在建模方法中,常见的有晶格构造、随机分布、从实验数据反演等。势参数获取途径则包括公开数据库(如ASE数据库)、专业文献、或通过第一性原理计算得到。
以金属Ni的拉伸模拟为例,用户需要定义Ni的原子类型,选择合适的EAM势,设定初始的晶体结构,然后设定拉伸载荷或应变速率,通过in文件控制模拟过程。模拟完成后,可以分析位错行为、应力-应变曲线等,以了解金属的塑性变形机制。
LAMMPS是一个强大的工具,对于想要提升在分子动力学模拟方面技能的初学者,理解其基本原理、掌握建模方法和in文件的编写至关重要。通过学习和实践,可以从LAMMPS的素人逐步成长为熟练的使用者。
剩余26页未读,继续阅读
相关推荐



m0_64178018
- 粉丝: 0
 我的内容管理
展开
我的内容管理
展开







最新资源
- 网页自动刷新工具 v1.1 - 自定义时间间隔与关机
- pt-1.4协程源码深度解析
- EP4CE6E22C8芯片三相正弦波发生器设计与实现
- 高效处理超大XML文件的查看工具介绍
- 64K极限挑战:国际程序设计大赛优秀3D作品展
- ENVI软件全面应用教程指南
- 学生档案管理系统设计与开发
- 网络伪书:社区驱动的在线音乐制图平台
- Lettuce 5.0.3中文API文档完整包下载指南
- 雅虎通Yahoo! Messenger v0.8.115即时聊天功能详解
- 将Android手机转变为IP监控摄像机
- PLSQL入门教程:变量声明与程序交互
- 掌握.NET三层架构:实例学习与源码解析
- WPF中Devexpress GridControl分组功能实例分析
- H3Viewer: VS2010专用高效帮助文档查看工具
- STM32CubeMX LED与按键初始化及外部中断处理教程
