福尔马林固定组织中基因组空间表达谱分析技术
PDF格式 | 1.94MB |
更新于2025-01-16
| 55 浏览量 | 举报
技术
福尔马林固定组织中全基因组空间表达谱分
析
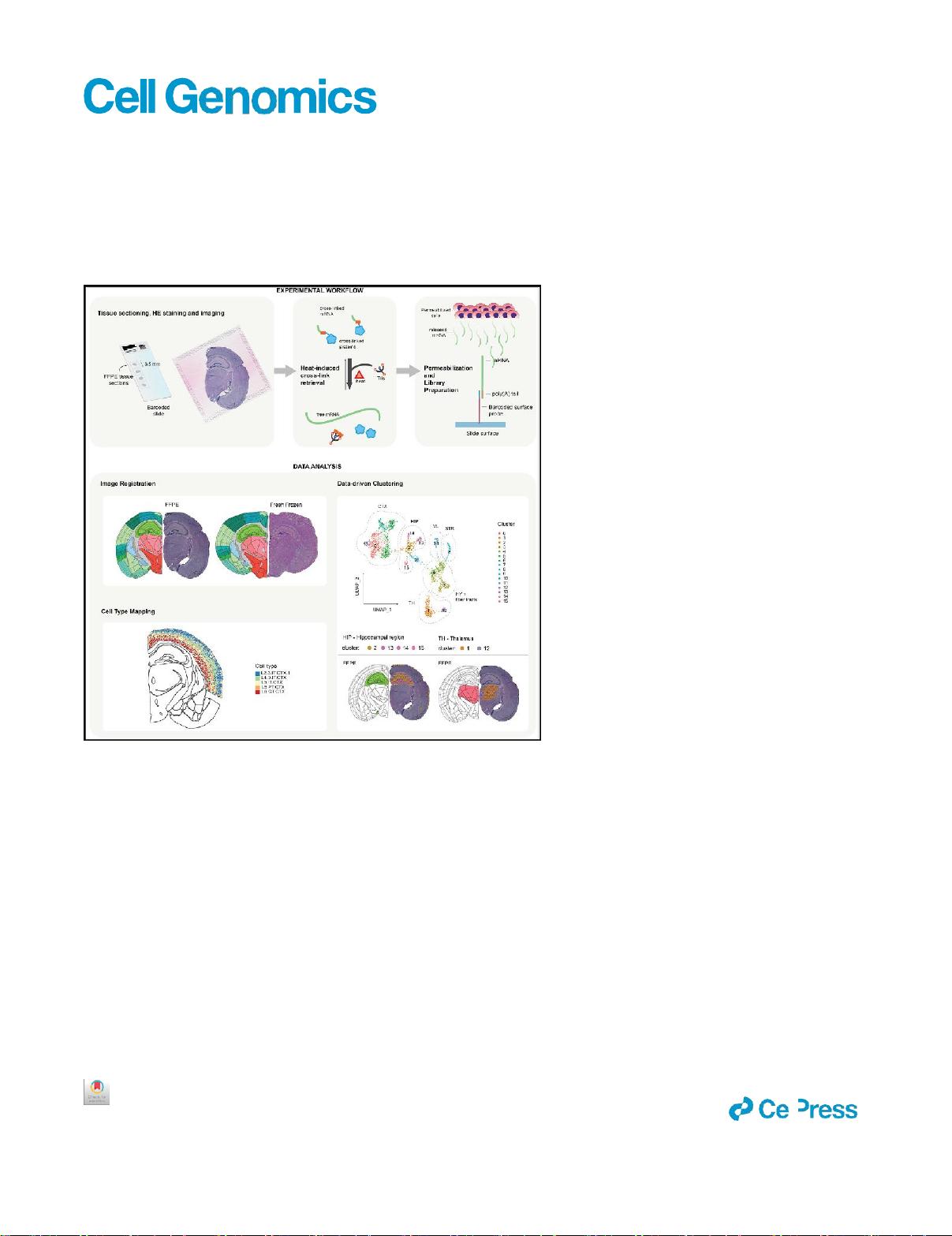
图形摘要
亮点
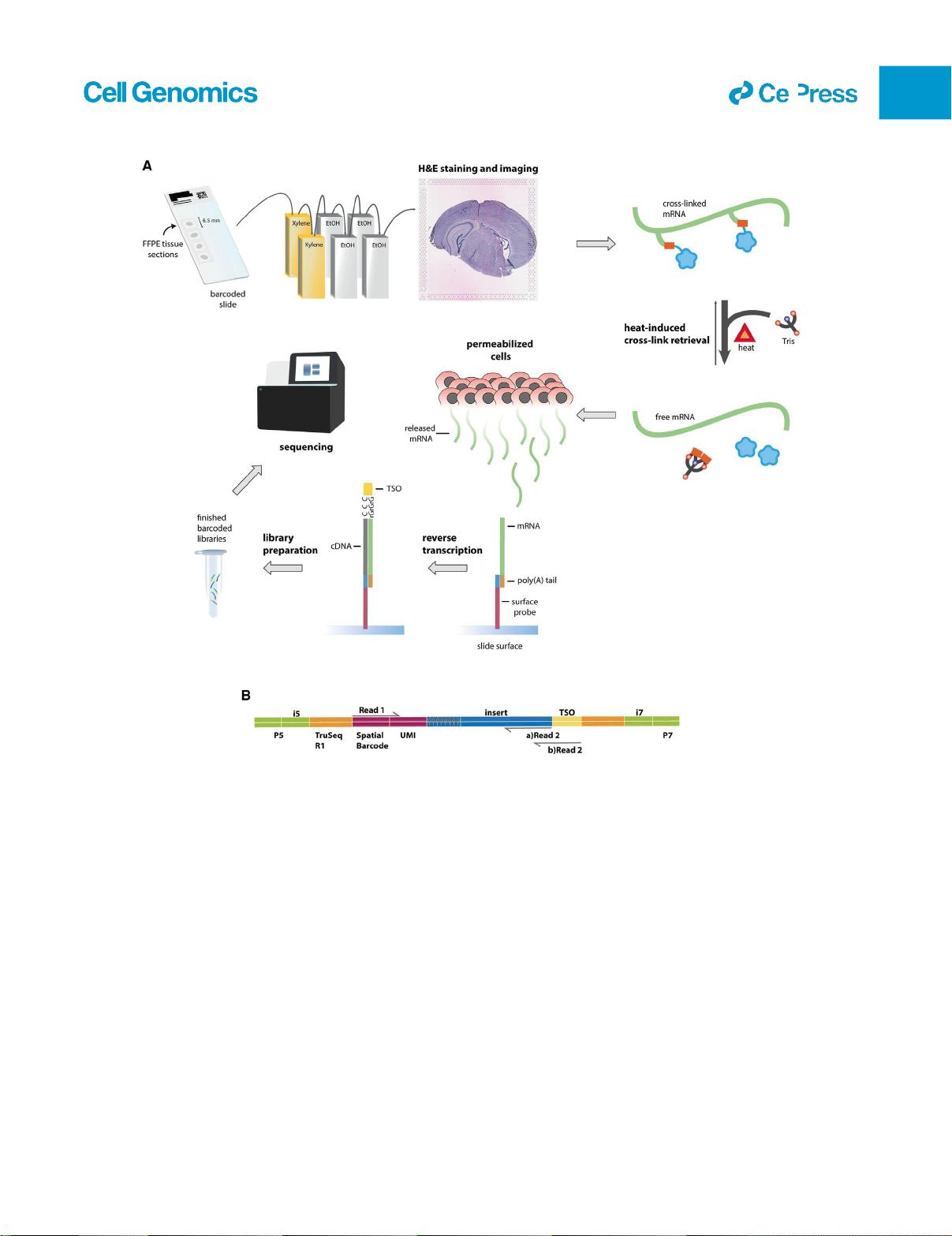
福尔马林固定组织的解交联使得能够进行全转录组
mRNA
分析
d
FFPE
小鼠脑数据的分子概况概括了已知的解剖结构
d
分子标记与卵巢癌
d
对
SARS-CoV-2
感染肺部的分析揭示了感染后的免疫反应
作者
Eva Gracia Villacampa,Ludvig
Larsson,Reza Mirzazadeh,.,Ali
Mirazimi,Joseph Carlson,Joakim
Lundeberg
对应
joakim. scilifelab.se
在 简介
Gracia Villacampa et al.
开发一种方法,
使用空间条形码载玻片并基于寡聚
(
dT
)
mRNA
捕获,对
FFPE
和
PFA
固定
的组织切片中的
mRNA
进行空间分析。
Gracia Villacampa等人,2021,细胞基因组学1,100065
2021
年
12
月
8
日
-
作者。
https://doi.org/10.1016/j.xgen.2021.100065
会



剩余24页未读,继续阅读
相关推荐


cpongm
- 粉丝: 6
 我的内容管理
展开
我的内容管理
展开







最新资源
- VB通过Modbus协议控制三菱PLC通讯实操指南
- simfinapi:R语言中简化SimFin数据获取与分析的包
- LabVIEW温度控制上位机程序开发指南
- 西门子工业网络通信实例解析与CP243-1应用
- 清华紫光全能王V9.1软件深度体验与功能解析
- VB实现Access数据库数据同步操作指南
- VB实现MSChart绘制实时监控曲线
- VC6.0通过实例深入访问Excel文件技巧
- 自动机可视化工具:编程语言与正则表达式的图形化解释
- 赛义德·莫比尼:揭秘其开创性技术成果
- 微信小程序开发教程:如何实现模仿ofo共享单车应用
- TrueTable在Windows10 64位及CAD2007中的完美适配
- 图解Win7搭建IIS7+PHP+MySQL+phpMyAdmin教程
- C#与LabVIEW联合采集NI设备的电压电流信号并创建Excel文件
- LP1800-3最小系统官方资料压缩包
- Linksys WUSB54GG无线网卡驱动程序下载指南
