Fiscore:R语言蛋白质结构分析与可视化工具
114 浏览量
更新于2025-01-16
收藏 2MB PDF 举报
"Fiscore是一款专为蛋白质结构数据可视化和探索设计的R语言软件包,旨在提供一种集成且用户友好的方式来评估蛋白质的构象和拓扑特性。它基于概率机器学习技术,特别是高斯混合建模,使得非专业人士也能方便地应用这些方法。Fiscore的核心在于其Fi-score方程,该方程考虑了蛋白质二面角和B因子,对于理解和解析复杂生物现象,以及在药物发现、目标评估和数据库构建中具有重要价值。该软件包还提供了一个交互式环境,便于研究人员探索多种功能。Fiscore v.0.1.3版本可通过CRAN和Github免费获取。其开发目的是为了支持结构生物信息学家的工作,同时也为非专业领域的研究者提供工具,帮助他们处理蛋白质结构数据。"
Fiscore软件包的核心功能包括:
1. **蛋白质结构特征评估**:Fiscore能够快速分析蛋白质的构象和拓扑特性,这对于理解蛋白质功能至关重要。
2. ** Fi-score方程**:该方程结合了蛋白质的二面角和B因子信息,有助于量化氨基酸残基的结构影响。
3. **高斯混合建模**:通过这一机器学习方法,Fiscore使得非专家也能理解和应用复杂的模型来处理蛋白质数据。
4. **交互式环境**:软件包提供了一个用户友好的界面,研究人员可以在这里探索和分析感兴趣的函数。
5. **生物制剂开发**:在抗体工程等生物制剂开发中,Fiscore可以帮助识别和表征潜在的结合位点,促进治疗剂的设计。
6. **目标分类与信息存储**:结构数据的得分形式可用于目标分类,存储目标配体信息,以及用于筛选研究。
7. **机器学习集成**:Fiscore的数据转换功能使其能够被集成到机器学习管道中,增强了数据分析的深度和广度。
在生命科学领域,尤其是蛋白质研究中,Fiscore软件包的引入极大地简化了数据分析过程,并促进了新疗法的开发。B因子,作为蛋白质结构中的一个重要参数,反映了原子的热运动和构象变化,对于理解和预测蛋白质的动态行为至关重要。通过Fiscore,研究人员能够更有效地利用这些信息,推动生命科学和药学院的研究进展。
A.
水磷铝石
生命科学中的人工智能
1
(
2021
)
100016
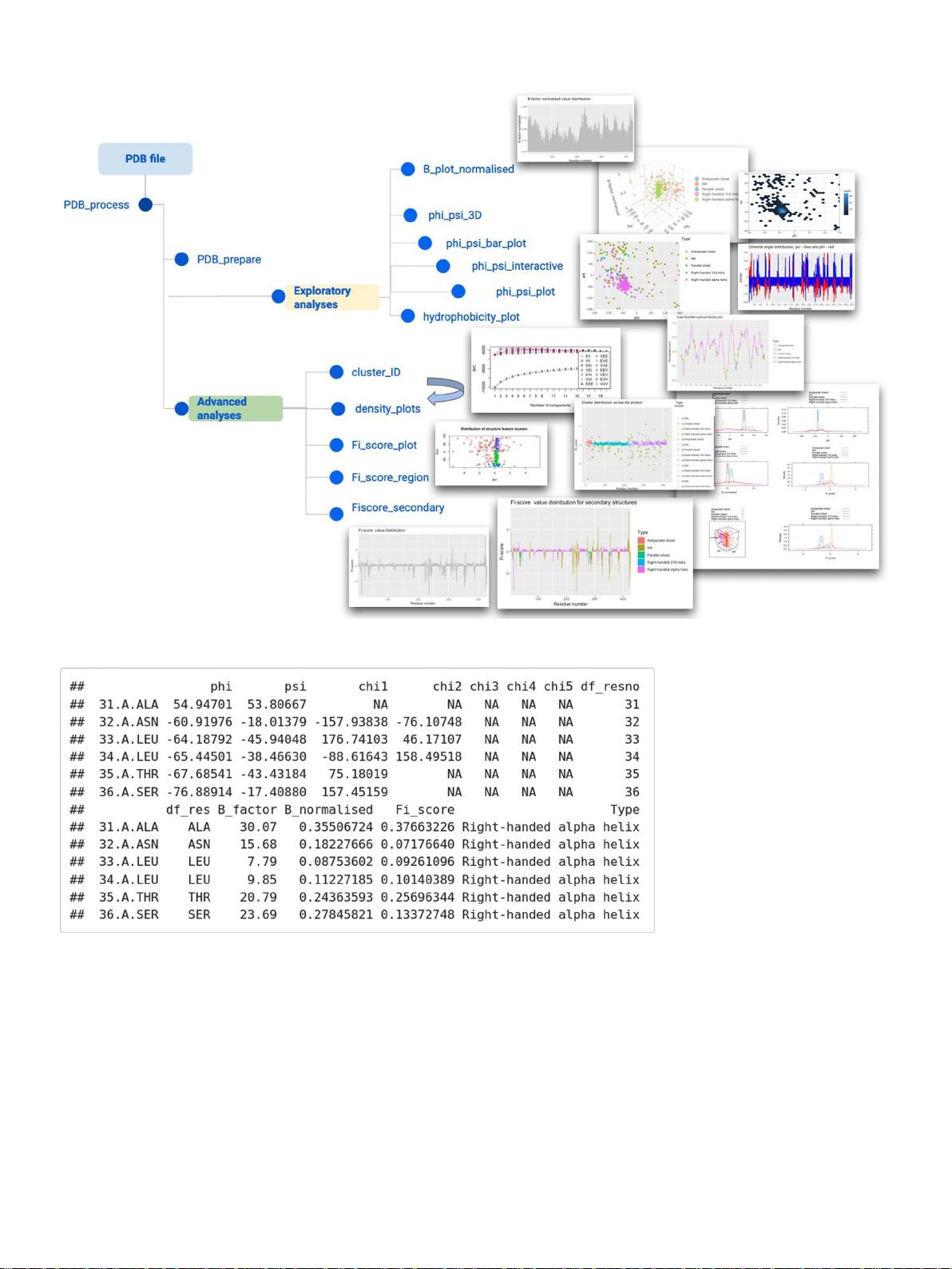
图1.一、包 装
特征的示意图可视化。
图二.
文件处理输出。
通过简单的函数调用访问基本分析,以探索二面角和因子的分
布方式。这些分析使关键参数的交互式和简单可视化变得更加容易,
而 这 些 参 数 目 前还 没有 在任 何其 他软 件包 中提 供。 例如 ,虽 然
具有许多用于探索文件的有用功能,但类似地,其他免
费提供 的工具 (作为 在线 服务分 发), 如 ,
,仅提供一维评估,不包含结构特征,也不处理文件。
然
而
, 在 简 单 的 函 数 调 用 中 结 合 了 序 列 、 结 构 和
物 理 化 学 分 析 , 以 快 速 探 索 用 户 数 据 。
调用 函数
()
二面角分布的可视化 ()
图值可视化()
通过分布
剩余10页未读,继续阅读
1736 浏览量
454 浏览量
2178 浏览量
点击了解资源详情
171 浏览量
220 浏览量
145 浏览量

cpongm
- 粉丝: 6
 我的内容管理
展开
我的内容管理
展开







最新资源
- A7Demo.appstudio:探索JavaScript应用开发
- 百度地图范围内的标注点技术实现
- Foobar2000绿色汉化版:全面提升音频播放体验
- Rhythm Core .NET库:字符串与集合扩展方法详解
- 深入了解Tomcat源码及其依赖包结构
- 物流节约里程法的文档整理与实践分享
- NUnit3.vsix:快速安装NUnit三件套到VS2017及以上版本
- JQuery核心函数使用速查手册详解
- 多种风格的Select下拉框美化插件及其js代码下载
- Mac用户必备:SmartSVN版本控制工具介绍
- ELTE IK Web编程与Web开发课程内容详解
- QuartusII环境下的Verilog锁相环实现
- 横版过关游戏完整VC源码及资源包
- MVC后台管理框架2021版:源码与代码生成器详解
- 宗成庆主讲的自然语言理解课程PPT解析
- Memcached与Tomcat会话共享与Kryo序列化配置指南
