"韩国生物库GWAS: 72,298个体的76种性状研究及122个新关联"
79 浏览量
更新于2025-01-16
收藏 878KB PDF 举报
文章
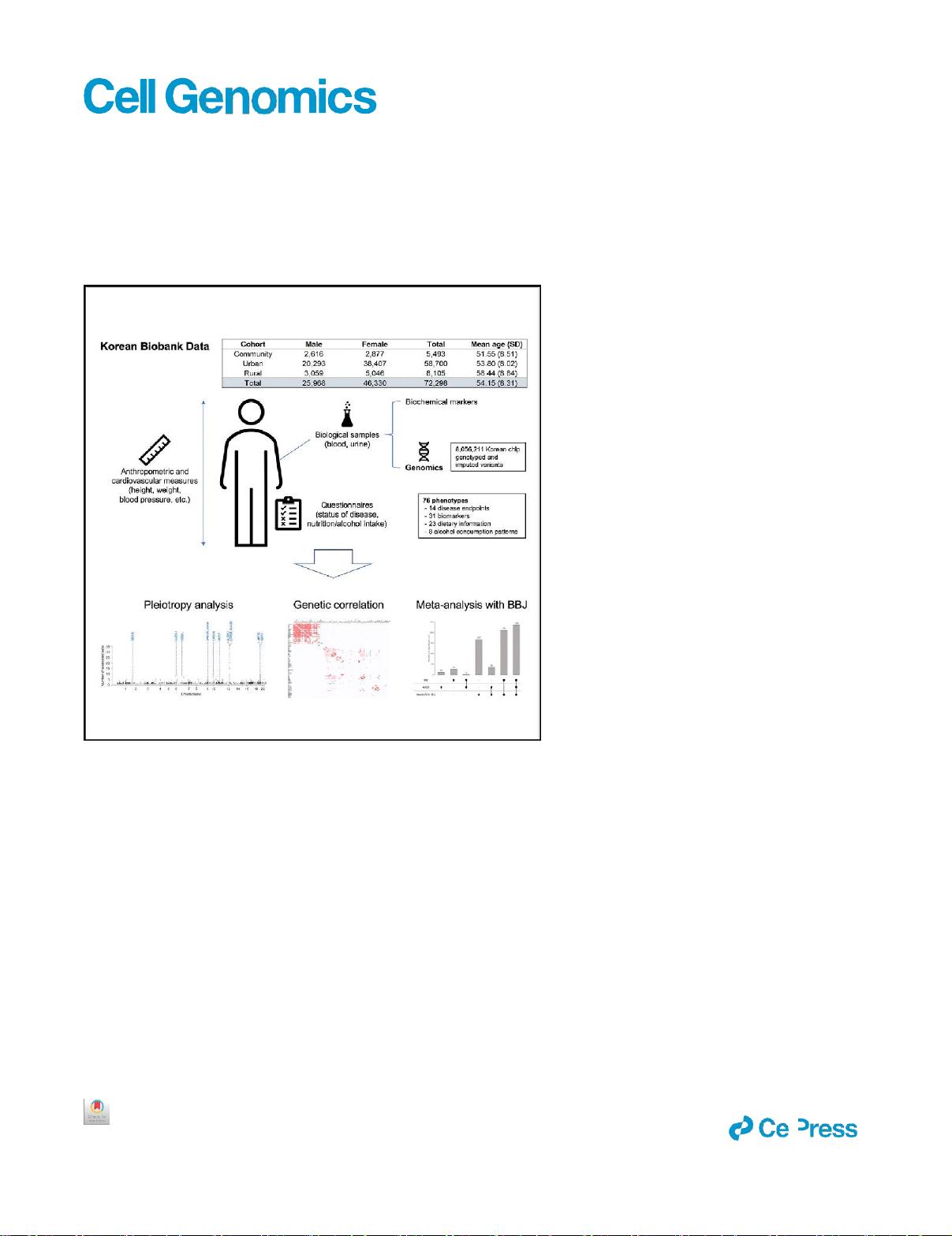
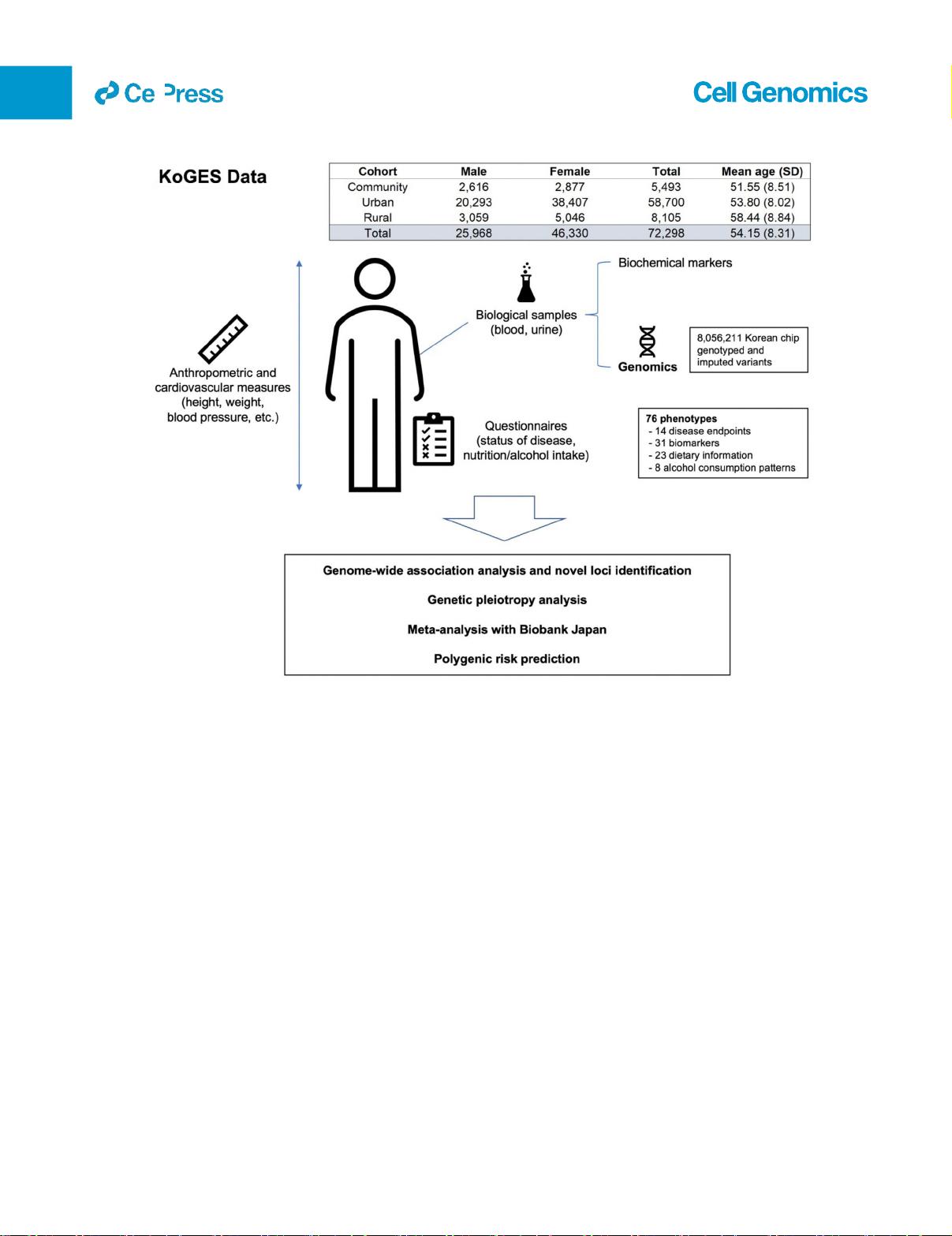
对韩国生物库数据中
72
,
298
个个体的
76
个性状进
行全基因组研究
图形摘要
亮点
d
韩国生物库中个体的
GWAS
确定了
122
种新的关联
d
多效基因和性状
d
一项包括韩国
GWAS
的荟萃分析提高了东亚人的风险预测准
确性
作者
Kisung Nam,Jangho Kim,Seunggeun Lee
对应
lee7801@snu.ac.kr
简言之
缺乏对不同祖先群体的全基因组关联研究
(
GWAS
),导致遗传发现不足。
Nam
等报告了韩国人
76
个性状的
GWAS
,确
定了
122
个新的关联,并通过荟萃分析证
明了多基因风险评分的预测性能的改善。
Nam等人,2022,细胞基因组学2,100189
2022
年
10
月
12
日
-
作者。
https://doi.org/10.1016/j.xgen.2022.100189
会

剩余13页未读,继续阅读
745 浏览量
133 浏览量
2230 浏览量
852 浏览量
149 浏览量
241 浏览量
387 浏览量
426 浏览量
138 浏览量

cpongm
- 粉丝: 6
 我的内容管理
展开
我的内容管理
展开







最新资源
- A7Demo.appstudio:探索JavaScript应用开发
- 百度地图范围内的标注点技术实现
- Foobar2000绿色汉化版:全面提升音频播放体验
- Rhythm Core .NET库:字符串与集合扩展方法详解
- 深入了解Tomcat源码及其依赖包结构
- 物流节约里程法的文档整理与实践分享
- NUnit3.vsix:快速安装NUnit三件套到VS2017及以上版本
- JQuery核心函数使用速查手册详解
- 多种风格的Select下拉框美化插件及其js代码下载
- Mac用户必备:SmartSVN版本控制工具介绍
- ELTE IK Web编程与Web开发课程内容详解
- QuartusII环境下的Verilog锁相环实现
- 横版过关游戏完整VC源码及资源包
- MVC后台管理框架2021版:源码与代码生成器详解
- 宗成庆主讲的自然语言理解课程PPT解析
- Memcached与Tomcat会话共享与Kryo序列化配置指南
