R语言ssGSEA代码
时间: 2023-07-10 11:14:15 浏览: 923
以下是基于R语言实现的ssGSEA富集分析代码:
```R
library(GSEABase)
library(ggplot2)
library(dplyr)
# 定义ssGSEA函数
ssgsea <- function(gene_exp, gene_sets, nperm=1000, weighted_score_type=1, permutation=TRUE,
min_size=1, max_size=5000, verbose=FALSE, seed=NULL) {
# 设置随机数种子
if (!is.null(seed)) set.seed(seed)
# 准备基因表达矩阵
gene_exp <- as.matrix(gene_exp)
# 准备基因集列表
gene_sets <- gene_sets[gene_sets$Size >= min_size & gene_sets$Size <= max_size, ]
# 计算基因集得分
pathways <- list()
for (i in 1:nrow(gene_sets)) {
# 获取基因集中的基因
genes <- gene_sets$genes[[i]]
# 计算每个样本的基因集得分
gss <- apply(gene_exp, 1, function(x) {
# 获取基因集中基因的表达值
pathway_exp <- x[which(names(x) %in% genes)]
# 计算基因集得分
if (weighted_score_type == 0) sum(pathway_exp)
else if (weighted_score_type == 1) mean(pathway_exp)
else if (weighted_score_type == -1) mean(abs(pathway_exp))
})
# 计算富集得分和p值
if (permutation) {
null_gss <- replicate(nperm, {
# 打乱基因表达值
shuffle_exp <- apply(gene_exp, 2, sample)
# 计算每个样本的基因集得分
apply(shuffle_exp, 1, function(x) {
# 获取基因集中基因的表达值
pathway_exp <- x[which(names(x) %in% genes)]
# 计算基因集得分
if (weighted_score_type == 0) sum(pathway_exp)
else if (weighted_score_type == 1) mean(pathway_exp)
else if (weighted_score_type == -1) mean(abs(pathway_exp))
})
})
null_gss <- apply(null_gss, 2, function(x) sort(x))
null_es <- apply(null_gss, 2, function(x) (mean(x > gss) - mean(x < gss)) / sd(x))
es <- (mean(gss) - mean(null_es)) / sd(null_es)
pval <- mean(null_es < mean(gss))
} else {
es <- (mean(gss) - mean(gss)) / sd(gss)
pval <- 1 - pnorm(es)
}
pathway <- gene_sets$Pathway[i]
pathways[[pathway]] <- list(es=es, pval=pval)
if (verbose) {
cat(sprintf("%s: ES = %.3f, p-value = %.3f\n", pathway, es, pval))
}
}
return(pathways)
}
# 使用示例
# 准备基因表达矩阵和基因集列表
data(geneExp)
data(geneSets)
# 进行ssGSEA富集分析
pathways <- ssgsea(gene_exp=geneExp, gene_sets=geneSets)
# 将结果转换为数据框并排序
pathways_df <- lapply(pathways, function(x) {
data.frame(Pathway=names(x), ES=x$es, pval=x$pval)
}) %>% do.call(rbind, .)
pathways_df <- pathways_df[order(pathways_df$pval), ]
# 绘制p值分布图
ggplot(pathways_df, aes(x=pval)) +
geom_density() +
xlab("p-value") +
ggtitle("Distribution of ssGSEA p-values")
```
该代码使用了GSEABase、ggplot2和dplyr等库进行计算和绘图。在使用时,需要将基因表达矩阵和基因集列表传递给`ssgsea`函数。此外,还可以设置其他参数,例如是否进行置换和置换次数等。函数返回一个包含富集分析结果的列表。可以将结果转换为数据框并排序,然后绘制p值分布图。
阅读全文
相关推荐
大家在看
QT实现动画右下角提示信息弹窗
QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动画右下角提示信息弹窗QT实现动
【瑞幸财报下载】2017-2023年Q1瑞幸咖啡财报LK.O年报财务报表数据Excel招股书中文下载
瑞幸咖啡 LK.O(退市);
2017-2023年Q1;
格式:财报Excel/
招股书PDF/年报PDF;
立即下载:
部分截图
1.三大财务报表Excel:
资产负债表>>
利润表>>
现金流量表>>
2.财务分析比率指标Excel:
3
.招股说明书PDF:
C语言课程设计《校园新闻发布管理系统》.zip
C语言课程设计《校园新闻发布管理系统》.zip C语言课程设计《校园新闻发布管理系统》.zip C语言课程设计《校园新闻发布管理系统》.zip C语言课程设计《校园新闻发布管理系统》.zip C语言课程设计《校园新闻发布管理系统》.zip C语言课程设计《校园新闻发布管理系统》.zip C语言课程设计《校园新闻发布管理系统》.zip C语言课程设计《校园新闻发布管理系统》.zip C语言课程设计《校园新闻发布管理系统》.zip C语言课程设计《校园新闻发布管理系统》.zip C语言课程设计《校园新闻发布管理系统》.zip C语言课程设计《校园新闻发布管理系统》.zip C语言课程设计《校园新闻发布管理系统》.zip C语言课程设计《校园新闻发布管理系统》.zip C语言课程设计《校园新闻发布管理系统》.zi
项目资源具有较高的学习借鉴价值,也可直接拿来修改复现。可以在这些基础上学习借鉴进行修改和扩展,实现其它功能。
可下载学习借鉴,你会有所收获。
# 注意 1. 本资源仅用于开源学习和技术交流。不可商用等,一切后果由使用者承担。2. 部分字体以及插图等来自网络,若是侵权请联系删除。
测量变频损耗L的方框图如图-所示。-微波电路实验讲义
测量变频损耗L的方框图如图1-1所示。
图1-1 实验线路
实验线路连接
本振源
信号源
功率计
定向耦合器
超高频毫伏表
滤波器 50Ω
混频器
毫安表
冲击波在水深方向传播规律数值仿真研究模型文件
以1000m水深为例,给出了TNT球形装药水下爆炸冲击波载荷在水深方向传播数值仿真研究的模型文件
最新推荐
S变换+Sockwell R G , Mansinha L , Lowe R P . Localization of the complex spectrum: the S transformJ
s变换用的高斯窗函数( 高斯窗是指数窗的一种,它也无负的旁瓣,而且没有旁瓣波动,因而不回引起计算谱中假的极大值或极小值,而且高斯窗频率窗函数的主瓣比指数窗的主瓣窄,分辨率比指数窗有所提高。
2021科大讯飞车辆贷违预测大赛冠军源码+全部资料.zip
2021科大讯飞车辆贷违预测大赛冠军源码+全部资料.zip
[资源说明]
1、该项目是团队成员近期最新开发,代码完整,资料齐全,含设计文档等
2、上传的项目源码经过严格测试,功能完善且能正常运行,请放心下载使用!
3、本项目适合计算机相关专业(人工智能、通信工程、自动化、电子信息、物联网等)的高校学生、教师、科研工作者、行业从业者下载使用,可借鉴学习,也可直接作为毕业设计、课程设计、作业、项目初期立项演示等,也适合小白学习进阶,遇到问题不懂就问,欢迎交流。
4、如果基础还行,可以在此代码基础上进行修改,以实现其他功能,也可直接用于毕设、课设、作业等。
5、不懂配置和运行,可远程教学
欢迎下载,学习使用!
AI图像处理工具包-一键抠图、背景切换、旧照片修复、人像漫画化、视频卡通化(Python+OpenCV+Dlib+TensorFlow).zip
AI图像处理工具包-一键抠图、背景切换、旧照片修复、人像漫画化、视频卡通化(Python+OpenCV+Dlib+TensorFlow).zip
[资源说明]
1、该项目是团队成员近期最新开发,代码完整,资料齐全,含设计文档等
2、上传的项目源码经过严格测试,功能完善且能正常运行,请放心下载使用!
3、本项目适合计算机相关专业(人工智能、通信工程、自动化、电子信息、物联网等)的高校学生、教师、科研工作者、行业从业者下载使用,可借鉴学习,也可直接作为毕业设计、课程设计、作业、项目初期立项演示等,也适合小白学习进阶,遇到问题不懂就问,欢迎交流。
4、如果基础还行,可以在此代码基础上进行修改,以实现其他功能,也可直接用于毕设、课设、作业等。
5、不懂配置和运行,可远程教学
欢迎下载,学习使用!
基于java+springboot+vue+mysql的远程教育网站设计与实现.docx
基于java+springboot+vue+mysql的远程教育网站设计与实现.docx
springboot005学生心理咨询评估系统(源码+数据库+论文+PPT+包调试+一对一指导)
毕业设计资料,计算机毕业设计,源码,毕业论文,毕业答辩,答辩PPT,Java毕业设计,php毕业设计,ASP.NET毕业设计,毕业指导,计算机作业,php作业,java作业,ASP.NET作业,编程作业,管理系统,网站,app,毕业设计学习,Java学习,php学习,ASP.NET学习,java课程,php课程,ASP.NET课程,答辩技巧,SQLSERVER数据库,Mysql数据库,jdbc,SSM框架,SpringBoot框架,Html5,小程序
HTML挑战:30天技术学习之旅
资源摘要信息: "desafio-30dias"
标题 "desafio-30dias" 暗示这可能是一个与挑战或训练相关的项目,这在编程和学习新技能的上下文中相当常见。标题中的数字“30”很可能表明这个挑战涉及为期30天的时间框架。此外,由于标题是西班牙语,我们可以推测这个项目可能起源于或至少是针对西班牙语使用者的社区。标题本身没有透露技术上的具体内容,但挑战通常涉及一系列任务,旨在提升个人的某项技能或知识水平。
描述 "desafio-30dias" 并没有提供进一步的信息,它重复了标题的内容。因此,我们不能从中获得关于项目具体细节的额外信息。描述通常用于详细说明项目的性质、目标和期望成果,但由于这里没有具体描述,我们只能依靠标题和相关标签进行推测。
标签 "HTML" 表明这个挑战很可能与HTML(超文本标记语言)有关。HTML是构成网页和网页应用基础的标记语言,用于创建和定义内容的结构、格式和语义。由于标签指定了HTML,我们可以合理假设这个30天挑战的目的是学习或提升HTML技能。它可能包含创建网页、实现网页设计、理解HTML5的新特性等方面的任务。
压缩包子文件的文件名称列表 "desafio-30dias-master" 指向了一个可能包含挑战相关材料的压缩文件。文件名中的“master”表明这可能是一个主文件或包含最终版本材料的文件夹。通常,在版本控制系统如Git中,“master”分支代表项目的主分支,用于存放项目的稳定版本。考虑到这个文件名称的格式,它可能是一个包含所有相关文件和资源的ZIP或RAR压缩文件。
结合这些信息,我们可以推测,这个30天挑战可能涉及了一系列的编程任务和练习,旨在通过实践项目来提高对HTML的理解和应用能力。这些任务可能包括设计和开发静态和动态网页,学习如何使用HTML5增强网页的功能和用户体验,以及如何将HTML与CSS(层叠样式表)和JavaScript等其他技术结合,制作出丰富的交互式网站。
综上所述,这个项目可能是一个为期30天的HTML学习计划,设计给希望提升前端开发能力的开发者,尤其是那些对HTML基础和最新标准感兴趣的人。挑战可能包含了理论学习和实践练习,鼓励参与者通过构建实际项目来学习和巩固知识点。通过这样的学习过程,参与者可以提高在现代网页开发环境中的竞争力,为创建更加复杂和引人入胜的网页打下坚实的基础。
【CodeBlocks精通指南】:一步到位安装wxWidgets库(新手必备)

# 摘要
本文旨在为使用CodeBlocks和wxWidgets库的开发者提供详细的安装、配置、实践操作指南和性能优化建议。文章首先介绍了CodeBlocks和wxWidgets库的基本概念和安装流程,然后深入探讨了CodeBlocks的高级功能定制和wxWidgets的架构特性。随后,通过实践操作章节,指导读者如何创建和运行一个wxWidgets项目,包括界面设计、事件
andorid studio 配置ERROR: Cause: unable to find valid certification path to requested target
### 解决 Android Studio SSL 证书验证问题
当遇到 `unable to find valid certification path` 错误时,这通常意味着 Java 运行环境无法识别服务器提供的 SSL 证书。解决方案涉及更新本地的信任库或调整项目中的网络请求设置。
#### 方法一:安装自定义 CA 证书到 JDK 中
对于企业内部使用的私有 CA 颁发的证书,可以将其导入至 JRE 的信任库中:
1. 获取 `.crt` 或者 `.cer` 文件形式的企业根证书;
2. 使用命令行工具 keytool 将其加入 cacerts 文件内:
```
VC++实现文件顺序读写操作的技巧与实践
资源摘要信息:"vc++文件的顺序读写操作"
在计算机编程中,文件的顺序读写操作是最基础的操作之一,尤其在使用C++语言进行开发时,了解和掌握文件的顺序读写操作是十分重要的。在Microsoft的Visual C++(简称VC++)开发环境中,可以通过标准库中的文件操作函数来实现顺序读写功能。
### 文件顺序读写基础
顺序读写指的是从文件的开始处逐个读取或写入数据,直到文件结束。这与随机读写不同,后者可以任意位置读取或写入数据。顺序读写操作通常用于处理日志文件、文本文件等不需要频繁随机访问的文件。
### VC++中的文件流类
在VC++中,顺序读写操作主要使用的是C++标准库中的fstream类,包括ifstream(用于从文件中读取数据)和ofstream(用于向文件写入数据)两个类。这两个类都是从fstream类继承而来,提供了基本的文件操作功能。
### 实现文件顺序读写操作的步骤
1. **包含必要的头文件**:要进行文件操作,首先需要包含fstream头文件。
```cpp
#include <fstream>
```
2. **创建文件流对象**:创建ifstream或ofstream对象,用于打开文件。
```cpp
ifstream inFile("example.txt"); // 用于读操作
ofstream outFile("example.txt"); // 用于写操作
```
3. **打开文件**:使用文件流对象的成员函数open()来打开文件。如果不需要在创建对象时指定文件路径,也可以在对象创建后调用open()。
```cpp
inFile.open("example.txt", std::ios::in); // 以读模式打开
outFile.open("example.txt", std::ios::out); // 以写模式打开
```
4. **读写数据**:使用文件流对象的成员函数进行数据的读取或写入。对于读操作,可以使用 >> 运算符、get()、read()等方法;对于写操作,可以使用 << 运算符、write()等方法。
```cpp
// 读取操作示例
char c;
while (inFile >> c) {
// 处理读取的数据c
}
// 写入操作示例
const char *text = "Hello, World!";
outFile << text;
```
5. **关闭文件**:操作完成后,应关闭文件,释放资源。
```cpp
inFile.close();
outFile.close();
```
### 文件顺序读写的注意事项
- 在进行文件读写之前,需要确保文件确实存在,且程序有足够的权限对文件进行读写操作。
- 使用文件流进行读写时,应注意文件流的错误状态。例如,在读取完文件后,应检查文件流是否到达文件末尾(failbit)。
- 在写入文件时,如果目标文件不存在,某些open()操作会自动创建文件。如果文件已存在,open()操作则会清空原文件内容,除非使用了追加模式(std::ios::app)。
- 对于大文件的读写,应考虑内存使用情况,避免一次性读取过多数据导致内存溢出。
- 在程序结束前,应该关闭所有打开的文件流。虽然文件流对象的析构函数会自动关闭文件,但显式调用close()是一个好习惯。
### 常用的文件操作函数
- `open()`:打开文件。
- `close()`:关闭文件。
- `read()`:从文件读取数据到缓冲区。
- `write()`:向文件写入数据。
- `tellg()` 和 `tellp()`:分别返回当前读取位置和写入位置。
- `seekg()` 和 `seekp()`:设置文件流的位置。
### 总结
在VC++中实现顺序读写操作,是进行文件处理和数据持久化的基础。通过使用C++的标准库中的fstream类,我们可以方便地进行文件读写操作。掌握文件顺序读写不仅可以帮助我们在实际开发中处理数据文件,还可以加深我们对C++语言和文件I/O操作的理解。需要注意的是,在进行文件操作时,合理管理和异常处理是非常重要的,这有助于确保程序的健壮性和数据的安全。
【大数据时代必备:Hadoop框架深度解析】:掌握核心组件,开启数据科学之旅
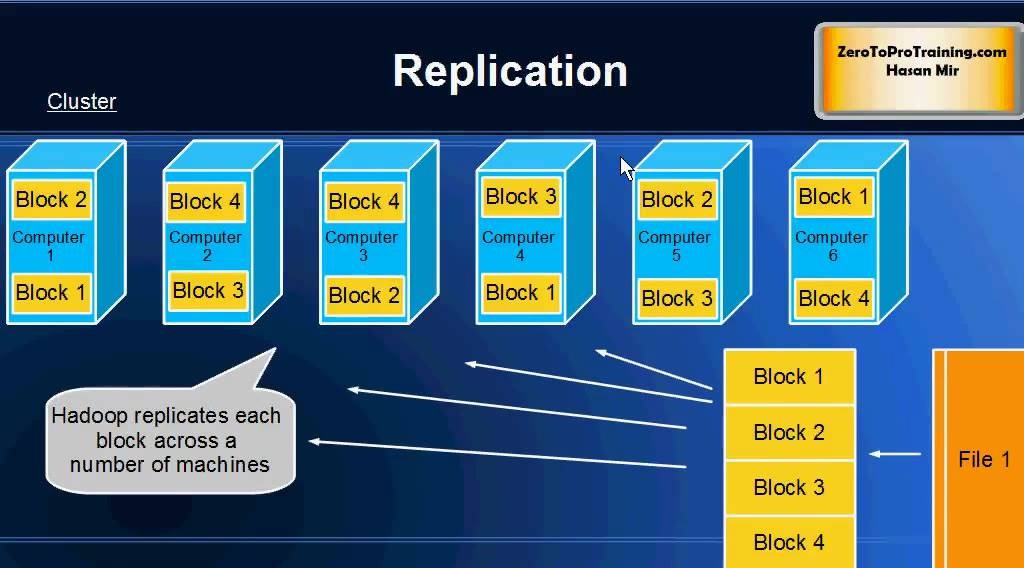
# 摘要
Hadoop作为一个开源的分布式存储和计算框架,在大数据处理领域发挥着举足轻重的作用。本文首先对Hadoop进行了概述,并介绍了其生态系统中的核心组件。深入分