lammps如何进行浸润测试
时间: 2023-11-01 15:02:48 浏览: 64
LAMMPS是一种经典的分子动力学模拟软件,可以用于模拟和研究材料的结构和性质。在进行浸润测试时,LAMMPS可以通过以下步骤来实现:
1. 创建模拟系统:首先需要定义一个模拟系统,包括表示固体材料和润滑剂的分子模型。可以使用现有的力场模型或根据实验数据自定义模型。
2. 设置原子组:将固体材料和润滑剂的原子按照相应的组织方式放置在模拟系统中。可以根据需要设置多个固体材料的区域和多个润滑剂的分子。
3. 定义相关参数:指定模拟系统的温度、压力、模拟时间和边界条件等参数。这些参数将影响模拟结果的准确性和稳定性。
4. 进行浸润模拟:设置固体材料表面的初始状态,例如,固体表面上是否存在表面分子层或斜面。然后,通过引入润滑剂分子到模拟系统中,模拟润滑剂在固体材料表面的运动,以研究浸润过程中的润滑效果。
5. 分析模拟结果:根据模拟的时间序列数据,可以计算浸润测试中的相关性质,如固体材料和润滑剂分子之间的相互作用力、润滑剂分子在固体表面的扩散行为等。
6. 优化参数:根据模拟结果,可以调整模型参数和条件,进一步优化模拟的准确性和稳定性。这可能需要多次迭代和调整模型,以获得更准确的浸润测试结果。
综上所述,LAMMPS通过建立模拟系统、定义参数、进行浸润模拟、分析结果和优化参数的步骤,可以进行浸润测试的模拟研究。这些模拟结果可以为浸润现象的机制、性质和应用提供有价值的信息。
相关问题
lammps力学性能测试数据的matlab处理应用
LAMMPS是一种常用的大分子动力学模拟软件,用于研究材料力学性能。在进行力学性能测试时,LAMMPS会产生大量的数据,包括原子坐标、原子速度、原子力等信息,这些数据可以通过使用Matlab进行处理和分析。
对于力学性能测试数据的处理,Matlab可以进行各种数据处理操作,包括数据读取、数据筛选、数据变换以及数据可视化等。首先,可以使用Matlab的读取函数,如`importdata`或`readtable`将LAMMPS输出的数据文件导入到Matlab中,可以得到一个矩阵或表格型的数据结构。
在掌握了数据后,可以使用Matlab提供的各种工具对数据进行筛选和处理。例如,可以根据需要选择特定的原子类型或区域,从原始数据中筛选出感兴趣的数据。可以使用Matlab的条件语句和循环语句对数据进行操作,如计算力的大小、原子的受力方向等。
Matlab还提供了丰富的数学函数和工具箱,可用于对力学性能数据进行数值计算和分析。例如,可以计算应力张量、应变张量,并根据得到的结果评估材料的力学性能。Matlab的统计工具箱还可以用于对数据的统计分析,如计算平均值、方差、相关系数等。
最后,Matlab还提供了强大的数据可视化功能,可以绘制各种图形以展示力学性能数据的特征。例如,可以绘制原子位置的散点图、力的分布图、应力-应变曲线等,帮助研究人员更直观地理解和分析力学性能测试数据。
综上所述,Matlab在LAMMPS力学性能测试数据的处理中具有广泛的应用,可以帮助研究人员快速获取、处理和分析数据,从而更深入地研究材料的力学性能。
请使用lammps语言来进行编程
以下是使用LAMMPS语言进行分子动力学模拟三维梯度纳米材料的一个示例:
```
# 初始化
units lj
dimension 3
boundary p p p
atom_style atomic
read_data data.gradient
# 定义势函数
pair_style lj/cut 2.5
pair_coeff * * 1.0 1.0 2.5
variable ecoh equal -4.0
variable r0 equal 2.5
variable alpha equal 1.0
variable rmin equal {r0}/pow(2,1/6)
variable rmax equal {rmin}*{alpha}
variable rc equal {rmax}+1.0
neighbor 2.0 bin
neigh_modify every 1 delay 0 check yes
# 定义梯度势函数
variable Uc1 atom -((x-5.0)**2+(y-5.0)**2)/2.0
variable Uc2 atom -((x-5.0)**2+(y-5.0)**2)/2.0-((z-5.0)/4.0)**2
variable Uc3 atom -((x-5.0)**2+(y-5.0)**2)/2.0-((z-5.0)/2.0)**2
variable Uc4 atom -((x-5.0)**2+(y-5.0)**2)/2.0-((z-5.0)/1.0)**2
variable Uc5 atom -((x-5.0)**2+(y-5.0)**2)/2.0-((z-5.0)/0.5)**2
variable Uc6 atom -((x-5.0)**2+(y-5.0)**2)/2.0-((z-5.0)/0.25)**2
variable Uc7 atom -((x-5.0)**2+(y-5.0)**2)/2.0-((z-5.0)/0.125)**2
variable Uc8 atom -((x-5.0)**2+(y-5.0)**2)/2.0-((z-5.0)/0.0625)**2
variable Uc9 atom -((x-5.0)**2+(y-5.0)**2)/2.0-((z-5.0)/0.03125)**2
variable Uc10 atom -((x-5.0)**2+(y-5.0)**2)/2.0-((z-5.0)/0.015625)**2
variable Uc11 atom -((x-5.0)**2+(y-5.0)**2)/2.0-((z-5.0)/0.0078125)**2
variable Uc12 atom -((x-5.0)**2+(y-5.0)**2)/2.0-((z-5.0)/0.00390625)**2
variable Uc13 atom -((x-5.0)**2+(y-5.0)**2)/2.0-((z-5.0)/0.001953125)**2
variable Uc14 atom -((x-5.0)**2+(y-5.0)**2)/2.0-((z-5.0)/0.0009765625)**2
variable Uc15 atom -((x-5.0)**2+(y-5.0)**2)/2.0-((z-5.0)/0.00048828125)**2
variable Uc16 atom -((x-5.0)**2+(y-5.0)**2)/2.0-((z-5.0)/0.000244140625)**2
variable Uc17 atom -((x-5.0)**2+(y-5.0)**2)/2.0-((z-5.0)/0.0001220703125)**2
variable Uc18 atom -((x-5.0)**2+(y-5.0)**2)/2.0-((z-5.0)/0.00006103515625)**2
variable Uc19 atom -((x-5.0)**2+(y-5.0)**2)/2.0-((z-5.0)/0.000030517578125)**2
variable Uc20 atom -((x-5.0)**2+(y-5.0)**2)/2.0-((z-5.0)/0.0000152587890625)**2
pair_coeff * * v_Uc1
pair_coeff * * v_Uc2
pair_coeff * * v_Uc3
pair_coeff * * v_Uc4
pair_coeff * * v_Uc5
pair_coeff * * v_Uc6
pair_coeff * * v_Uc7
pair_coeff * * v_Uc8
pair_coeff * * v_Uc9
pair_coeff * * v_Uc10
pair_coeff * * v_Uc11
pair_coeff * * v_Uc12
pair_coeff * * v_Uc13
pair_coeff * * v_Uc14
pair_coeff * * v_Uc15
pair_coeff * * v_Uc16
pair_coeff * * v_Uc17
pair_coeff * * v_Uc18
pair_coeff * * v_Uc19
pair_coeff * * v_Uc20
# 定义时间演化
timestep 0.001
thermo 100
thermo_style custom step temp press pe ke etotal vol lx ly lz
fix 1 all npt temp 1.0 1.0 100.0 iso 0.0 0.0 1000.0
run 10000
```
该程序使用了LAMMPS语言,通过分子动力学模拟方法,模拟了三维梯度纳米材料的时间演化过程。程序中包括了读取数据文件、定义势函数、定义梯度势函数、定义时间演化等步骤。程序可以输出模拟过程中的温度、压力、能量等信息,并根据需要进行修改和扩充,实现更加复杂的梯度纳米材料模拟。
相关推荐
最新推荐
lammps实例3.pdf
LAMMPS(大型分子多尺度模拟)是一个广泛使用的开源分子动力学软件,适用于模拟从纳米到微米尺度的各种物质,包括软物质和固体物理系统。它的灵活性和高性能使其能够处理当前大多数的势能模型,这得益于其高度优化的...
lammps实例5.pdf
总结来说,LAMMPS实例5展示了如何利用该软件进行金属熔化与凝固的模拟,以及如何分析不同温度下的材料特性。通过模拟,不仅可以验证理论预测,还能探索和解释材料在极端条件下表现出的非平衡行为,如过热和过冷现象...
lammps实例2.pdf
为了获得稳定的结构,我们需要对系统进行能量最小化,这通常通过LAMMPS提供的两种方法之一实现:共轭梯度(cg)或简单的下降(sd)。在这个例子中,我们选择使用sd进行能量最小化。 LAMMPS的输入文件(in.vacancy)...
lammps实例1.pdf
LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)是一款功能强大的分子模拟软件,它支持多种势能模型,如Stillinger-Weber (SW)势,并且具有高效的编程架构,适应于模拟软物质和固体物理系统。...
lammps实例4.pdf
LAMMPS(Large-scale Atomic/Molecular ...LAMMPS提供的强大工具使得这些计算成为可能,即使对于复杂的系统,也能高效地进行模拟研究。通过这些实例,我们可以深入理解LAMMPS的工作原理及其在材料科学中的应用价值。
批量文件重命名神器:HaoZipRename使用技巧
资源摘要信息:"超实用的批量文件改名字小工具rename"
在进行文件管理时,经常会遇到需要对大量文件进行重命名的场景,以统一格式或适应特定的需求。此时,批量重命名工具成为了提高工作效率的得力助手。本资源聚焦于介绍一款名为“rename”的批量文件改名工具,它支持增删查改文件名,并能够方便地批量操作,从而极大地简化了文件管理流程。
### 知识点一:批量文件重命名的需求与场景
在日常工作中,无论是出于整理归档的目的还是为了符合特定的命名规则,批量重命名文件都是一个常见的需求。例如:
- 企业或组织中的文件归档,可能需要按照特定的格式命名,以便于管理和检索。
- 在处理下载的多媒体文件时,可能需要根据文件类型、日期或其他属性重新命名。
- 在软件开发过程中,对代码文件或资源文件进行统一的命名规范。
### 知识点二:rename工具的基本功能
rename工具专门设计用来处理文件名的批量修改,其基本功能包括但不限于:
- **批量修改**:一次性对多个文件进行重命名。
- **增删操作**:在文件名中添加或删除特定的文本。
- **查改功能**:查找文件名中的特定文本并将其替换为其他文本。
- **格式统一**:为一系列文件统一命名格式。
### 知识点三:使用rename工具的具体操作
以rename工具进行批量文件重命名通常遵循以下步骤:
1. 选择文件:根据需求选定需要重命名的文件列表。
2. 设定规则:定义重命名的规则,比如在文件名前添加“2023_”,或者将文件名中的“-”替换为“_”。
3. 执行重命名:应用设定的规则,批量修改文件名。
4. 预览与确认:在执行之前,工具通常会提供预览功能,允许用户查看重命名后的文件名,并进行最终确认。
### 知识点四:rename工具的使用场景
rename工具在不同的使用场景下能够发挥不同的作用:
- **IT行业**:对于软件开发者或系统管理员来说,批量重命名能够快速调整代码库中文件的命名结构,或者修改服务器上的文件名。
- **媒体制作**:视频编辑和摄影师经常需要批量重命名图片和视频文件,以便更好地进行分类和检索。
- **教育与学术**:教授和研究人员可能需要批量重命名大量的文档和资料,以符合学术规范或方便资料共享。
### 知识点五:rename工具的高级特性
除了基本的批量重命名功能,一些高级的rename工具可能还具备以下特性:
- **正则表达式支持**:利用正则表达式可以进行复杂的查找和替换操作。
- **模式匹配**:可以定义多种匹配模式,满足不同的重命名需求。
- **图形用户界面**:提供直观的操作界面,简化用户的操作流程。
- **命令行操作**:对于高级用户,可以通过命令行界面进行更为精准的定制化操作。
### 知识点六:与rename相似的其他批量文件重命名工具
除了rename工具之外,还有多种其他工具可以实现批量文件重命名的功能,如:
- **Bulk Rename Utility**:一个功能强大的批量重命名工具,特别适合Windows用户。
- **Advanced Renamer**:提供图形界面,并支持脚本,用户可以创建复杂的重命名方案。
- **MMB Free Batch Rename**:一款免费且易于使用的批量重命名工具,具有直观的用户界面。
### 知识点七:避免批量重命名中的常见错误
在使用批量重命名工具时,有几个常见的错误需要注意:
- **备份重要文件**:在批量重命名之前,确保对文件进行了备份,以防意外发生。
- **仔细检查规则**:设置好规则之后,一定要进行检查,确保规则的准确性,以免出现错误的命名。
- **逐步执行**:如果不确定规则的效果,可以先小批量试运行规则,确认无误后再批量执行。
- **避免使用通配符**:在没有充分理解通配符含义的情况下,不建议使用,以免误操作。
综上所述,批量文件改名工具rename是一个高效、便捷的解决方案,用于处理大量文件的重命名工作。通过掌握其使用方法和技巧,用户可以显著提升文件管理的效率,同时减少重复劳动,保持文件系统的整洁和有序。
管理建模和仿真的文件
管理Boualem Benatallah引用此版本:布阿利姆·贝纳塔拉。管理建模和仿真。约瑟夫-傅立叶大学-格勒诺布尔第一大学,1996年。法语。NNT:电话:00345357HAL ID:电话:00345357https://theses.hal.science/tel-003453572008年12月9日提交HAL是一个多学科的开放存取档案馆,用于存放和传播科学研究论文,无论它们是否被公开。论文可以来自法国或国外的教学和研究机构,也可以来自公共或私人研究中心。L’archive ouverte pluridisciplinaire
RestTemplate性能优化秘籍:提升API调用效率,打造极致响应速度
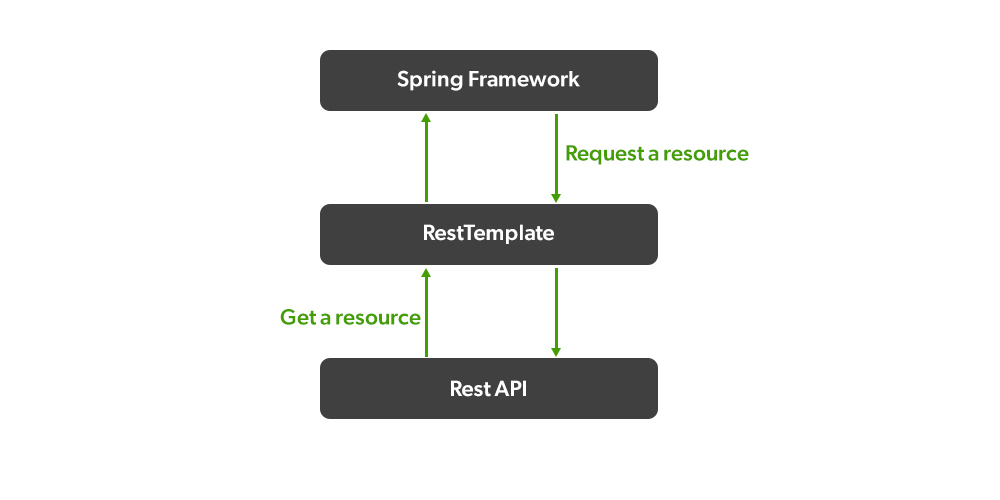
# 1. RestTemplate基础与API调用概述
## 1.1 RestTemplate简介
`RestTemplate`是Spring框架提供的一个同步HTTP客户端,设计用来简化客户端HTTP请求的发送以及HTTP响应的接收。它是针对Java开发者的REST API调用的高级抽象,为调用RESTful服务提供了便利。
Leader和follower 位置反了如何调换zookeeper
在ZooKeeper中,Leader和Follower的角色通常是静态分配的,并通过选举机制确定。如果需要调整它们的位置,通常是在集群初始化或者节点失效的情况下,会触发重新选举过程。
1. **停止服务**:首先,停止ZooKeeper服务的所有节点,包括当前的Leader和Follower。
2. **修改配置**:打开zoo.cfg配置文件,更改服务器列表(server.X=IP:port:角色),将原来的Leader的地址设为Follower,Follower的地址设为Leader。例如:
```
server.1=old_leader_ip:old_leader_po
简洁注册登录界面设计与代码实现
资源摘要信息:"在现代Web开发中,简洁美观的注册登录页面是用户界面设计的重要组成部分。简洁的页面设计不仅能够提升用户体验,还能提高用户完成注册或登录流程的意愿。本文将详细介绍如何创建两个简洁且功能完善的注册登录页面,涉及HTML5和前端技术。"
### 知识点一:HTML5基础
- **语义化标签**:HTML5引入了许多新标签,如`<header>`、`<footer>`、`<article>`、`<section>`等,这些语义化标签不仅有助于页面结构的清晰,还有利于搜索引擎优化(SEO)。
- **表单标签**:`<form>`标签是创建注册登录页面的核心,配合`<input>`、`<button>`、`<label>`等元素,可以构建出功能完善的表单。
- **增强型输入类型**:HTML5提供了多种新的输入类型,如`email`、`tel`、`number`等,这些类型可以提供更好的用户体验和数据校验。
### 知识点二:前端技术
- **CSS3**:简洁的页面设计往往需要巧妙的CSS布局和样式,如Flexbox或Grid布局技术可以实现灵活的页面布局,而CSS3的动画和过渡效果则可以提升交云体验。
- **JavaScript**:用于增加页面的动态功能,例如表单验证、响应式布局切换、与后端服务器交互等。
### 知识点三:响应式设计
- **媒体查询**:使用CSS媒体查询可以创建响应式设计,确保注册登录页面在不同设备上都能良好显示。
- **流式布局**:通过设置百分比宽度或视口单位(vw/vh),使得页面元素可以根据屏幕大小自动调整大小。
### 知识点四:注册登录页面设计细节
- **界面简洁性**:避免过多的装饰性元素,保持界面的整洁和专业感。
- **易用性**:设计简洁直观的用户交互,确保用户能够轻松理解和操作。
- **安全性和隐私**:注册登录页面应特别注意用户数据的安全,如使用HTTPS协议保护数据传输,以及在前端进行基本的输入验证。
### 知识点五:Zip文件内容解析
- **登录.zip**:该压缩包内可能包含登录页面的HTML、CSS和JavaScript文件,以及相关的图片和其他资源文件。开发者可以利用这些资源快速搭建一个登录界面。
- **滑动登录注册界面.zip**:该压缩包内可能包含了两个页面的文件,分别是注册和登录页面。文件可能包含用HTML5实现的滑动动画效果,通过CSS3和JavaScript的结合实现动态交互,提供更流畅的用户体验。
通过这些知识点,开发者能够创建出既简洁又功能完善的注册登录页面。需要注意的是,尽管页面设计要简洁,但安全措施不可忽视。使用加密技术保护用户数据,以及在用户端进行有效的数据验证,都是开发者在实现简洁界面同时需要考虑的安全要素。