在蛋白质组学实验中,如何利用胰酶消化的肽段数据进行数据库检索和比对,以准确鉴定蛋白质的身份?
时间: 2024-11-17 17:23:48 浏览: 11
在进行蛋白质组学实验时,胰酶消化是一种常见的肽段获取方法,用于将蛋白质分解成可检测的片段。为了鉴定这些肽段对应的蛋白质身份,研究人员通常会使用质谱分析技术,如Tandem MS(也称为MS/MS),来获得肽段的质量谱图。这些谱图包含了肽段的m/z(质量/电荷比)信息,是后续数据库检索和比对的关键数据。
参考资源链接:[蛋白质组学数据分析:Excel操作与软件应用详解](https://wenku.csdn.net/doc/kv9kqtphjy?spm=1055.2569.3001.10343)
在数据库检索和比对的过程中,研究人员会使用特定的蛋白质组学分析软件,如GPM(X!tandem)或TPP(The Trans-Proteomic Pipeline),将质谱分析得到的肽段质量谱图与蛋白质数据库中的理论肽段质量进行比对。比对的过程涉及到算法预测肽段的理论质量,并检查这些预测值与实验数据的一致性。通过这种比对,可以找到最匹配的蛋白质序列,进而确定原始蛋白质的身份。
为了帮助你更深入理解这一过程,建议参考《蛋白质组学数据分析:Excel操作与软件应用详解》。本书详细介绍了蛋白质组学数据分析的理论基础和实用技术,包括质谱分析技术、数据库检索以及如何使用各种工具软件进行肽段分析。通过本课程,你将学会如何处理实验数据,进行数据库比对,并最终鉴定出蛋白质的身份。掌握这些技能,将有助于你在蛋白质组学研究领域取得更深入的研究成果。
参考资源链接:[蛋白质组学数据分析:Excel操作与软件应用详解](https://wenku.csdn.net/doc/kv9kqtphjy?spm=1055.2569.3001.10343)
阅读全文
相关推荐
最新推荐
yolov5s nnie.zip
yolov5s nnieyolov5-nnieyolov5s nnieYOLOv5 pytorch -> onnx -> caffe -> .wk 1、模型是yolov5s,将focus层替换成stride为2的卷积层。reshape和permute层也做了调整。具体的修改过程可以参考这个大佬的文章https://blog.csdn.net/tangshopping/article/details/1100386052、模型是在hi3559av100上跑的,mapper版本是1.2。3、用法mkdir buildcd buildcmake -DCMAKE_TOOLCHAIN_FILE=../hi3559.toolchain.cmake ..make -j4./yolo_nnie参考https://blog.csdn.net/tangshopping/article/details/110038605watermelooon/nnie_yolohttps://github.com/ultralytics/yolov5https://githu
基于uni-app+uview-ui开发的校园云打印系统微信小程序项目源码+文档说明
基于uni-app+uview-ui开发的校园云打印系统微信小程序项目源码+文档说明,本资源中的源码都是经过本地编译过可运行的,评审分达到98分,资源项目的难度比较适中,内容都是经过助教老师审定过的能够满足学习、毕业设计、期末大作业和课程设计使用需求,如果有需要的话可以放心下载使用。
基于uni-app+uview-ui开发的校园云打印系统微信小程序项目源码+文档说明,本基于uni-app+uview-ui开发的校园云打印系统微信小程序项目源码+文档说明资源中的源码都是经过本地编译过可运行的,评审分达到98分,基于uni-app+uview-ui开发的校园云打印系统微信小程序项目源码+文档说明资源项目的难度比较适中,内容都是经过助教老师审定过的能够满足学习、毕业设计、期末大作业和课程设计使用需求,如果有需要的话可以放心下载使用。
本资源中的源码都是经过本地编译过可运行的,评审分达到98分,资源项目的难度比较适中,内容都是经过助教老师审定过的能够满足学习、毕业设计、期末大作业和课程设计使用需求,如果有需要的话可以放心下载使用。本资源中的源码都是经过本地编译过可运行的,评审分达到98分
使用Java写的一个简易的贪吃蛇小游戏.zip
使用Java写的一个简易的贪吃蛇小游戏.zip数据
计算机网络概述.docx
计算机网络概述概念:网络把主机连接起来,而互联网是把多种不同的网络连接起来,因此互联网是网络的网络。计算机网络主要包括三个部分:计算机(包括客户端、服务器)网络设备(路由器、交换机、防火墙等)传输介质(有线和无线)
ISP 互联网服务提供商ISP可以从互联网管理机构获得许多IP地址,同时拥有通信线路以及路由器等联网设备,个人或机构向ISP缴纳一定的费用就可以接入互联网。
目前的互联网是一种多层次ISP结构,ISP根据覆盖面积的大小分为主干ISP、地区ISP和本地ISP。互联网交换点IXP允许两个ISP直接相连而不用经过第三个ISP。
主机之间的通信方式 1、客户-服务器(C/S)
客户即是服务请求方,服务器是服务提供方。2、对等(P2P)
不区分客户和服务器
时延总时延=发送时延+传播时延+处理时延+排队时延计算机网络体系结构OSI:应用层、表示层、会话层、传输层、网络层、数据链路层、物理层五层协议:应用层、运输层、网络层、数据链路层、物理层TCP/IP:应用层、运输层、网际层、网络接口层
带通调制 模拟信号是连续的信号,数字信号是离散的信号。带通调制把数字信号转换为模拟信号。数据
数学建模学习资料 姜启源数学模型课件 M06 稳定性模型 共46页.pptx
数学建模学习资料 姜启源数学模型课件 M06 稳定性模型 共46页.pptx
JHU荣誉单变量微积分课程教案介绍
资源摘要信息:"jhu2017-18-honors-single-variable-calculus"
知识点一:荣誉单变量微积分课程介绍
本课程为JHU(约翰霍普金斯大学)的荣誉单变量微积分课程,主要针对在2018年秋季和2019年秋季两个学期开设。课程内容涵盖两个学期的微积分知识,包括整合和微分两大部分。该课程采用IBL(Inquiry-Based Learning)格式进行教学,即学生先自行解决问题,然后在学习过程中逐步掌握相关理论知识。
知识点二:IBL教学法
IBL教学法,即问题导向的学习方法,是一种以学生为中心的教学模式。在这种模式下,学生在教师的引导下,通过提出问题、解决问题来获取知识,从而培养学生的自主学习能力和问题解决能力。IBL教学法强调学生的主动参与和探索,教师的角色更多的是引导者和协助者。
知识点三:课程难度及学习方法
课程的第一次迭代主要包含问题,难度较大,学生需要有一定的数学基础和自学能力。第二次迭代则在第一次的基础上增加了更多的理论和解释,难度相对降低,更适合学生理解和学习。这种设计旨在帮助学生从实际问题出发,逐步深入理解微积分理论,提高学习效率。
知识点四:课程先决条件及学习建议
课程的先决条件为预演算,即在进入课程之前需要掌握一定的演算知识和技能。建议在使用这些笔记之前,先完成一些基础演算的入门课程,并进行一些数学证明的练习。这样可以更好地理解和掌握课程内容,提高学习效果。
知识点五:TeX格式文件
标签"TeX"意味着该课程的资料是以TeX格式保存和发布的。TeX是一种基于排版语言的格式,广泛应用于学术出版物的排版,特别是在数学、物理学和计算机科学领域。TeX格式的文件可以确保文档内容的准确性和排版的美观性,适合用于编写和分享复杂的科学和技术文档。
管理建模和仿真的文件
管理Boualem Benatallah引用此版本:布阿利姆·贝纳塔拉。管理建模和仿真。约瑟夫-傅立叶大学-格勒诺布尔第一大学,1996年。法语。NNT:电话:00345357HAL ID:电话:00345357https://theses.hal.science/tel-003453572008年12月9日提交HAL是一个多学科的开放存取档案馆,用于存放和传播科学研究论文,无论它们是否被公开。论文可以来自法国或国外的教学和研究机构,也可以来自公共或私人研究中心。L’archive ouverte pluridisciplinaire
【实战篇:自定义损失函数】:构建独特损失函数解决特定问题,优化模型性能
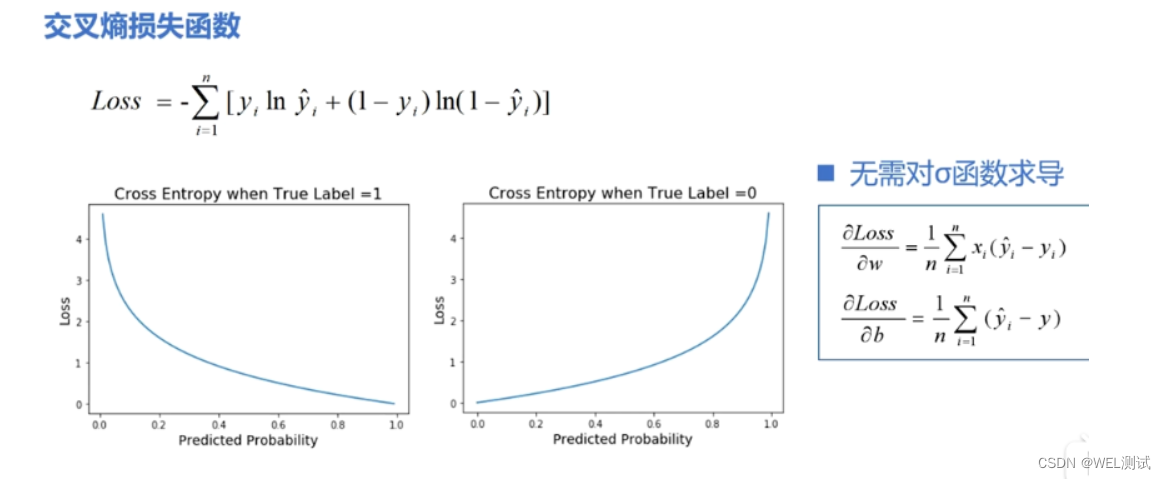
# 1. 损失函数的基本概念与作用
## 1.1 损失函数定义
损失函数是机器学习中的核心概念,用于衡量模型预测值与实际值之间的差异。它是优化算法调整模型参数以最小化的目标函数。
```math
L(y, f(x)) = \sum_{i=1}^{N} L_i(y_i, f(x_i))
```
其中,`L`表示损失函数,`y`为实际值,`f(x)`为模型预测值,`N`为样本数量,`L_i`为第`i`个样本的损失。
## 1.2 损
如何在ZYNQMP平台上配置TUSB1210 USB接口芯片以实现Host模式,并确保与Linux内核的兼容性?
要在ZYNQMP平台上实现TUSB1210 USB接口芯片的Host模式功能,并确保与Linux内核的兼容性,首先需要在硬件层面完成TUSB1210与ZYNQMP芯片的正确连接,保证USB2.0和USB3.0之间的硬件电路设计符合ZYNQMP的要求。
参考资源链接:[ZYNQMP USB主机模式实现与测试(TUSB1210)](https://wenku.csdn.net/doc/6nneek7zxw?spm=1055.2569.3001.10343)
具体步骤包括:
1. 在Vivado中设计硬件电路,配置USB接口相关的Bank502和Bank505引脚,同时确保USB时钟的正确配置。
Naruto爱好者必备CLI测试应用
资源摘要信息:"Are-you-a-Naruto-Fan:CLI测验应用程序,用于检查Naruto狂热者的知识"
该应用程序是一个基于命令行界面(CLI)的测验工具,设计用于测试用户对日本动漫《火影忍者》(Naruto)的知识水平。《火影忍者》是由岸本齐史创作的一部广受欢迎的漫画系列,后被改编成同名电视动画,并衍生出一系列相关的产品和文化现象。该动漫讲述了主角漩涡鸣人从忍者学校开始的成长故事,直到成为木叶隐村的领袖,期间包含了忍者文化、战斗、忍术、友情和忍者世界的政治斗争等元素。
这个测验应用程序的开发主要使用了JavaScript语言。JavaScript是一种广泛应用于前端开发的编程语言,它允许网页具有交互性,同时也可以在服务器端运行(如Node.js环境)。在这个CLI应用程序中,JavaScript被用来处理用户的输入,生成问题,并根据用户的回答来评估其对《火影忍者》的知识水平。
开发这样的测验应用程序可能涉及到以下知识点和技术:
1. **命令行界面(CLI)开发:** CLI应用程序是指用户通过命令行或终端与之交互的软件。在Web开发中,Node.js提供了一个运行JavaScript的环境,使得开发者可以使用JavaScript语言来创建服务器端应用程序和工具,包括CLI应用程序。CLI应用程序通常涉及到使用诸如 commander.js 或 yargs 等库来解析命令行参数和选项。
2. **JavaScript基础:** 开发CLI应用程序需要对JavaScript语言有扎实的理解,包括数据类型、函数、对象、数组、事件循环、异步编程等。
3. **知识库构建:** 测验应用程序的核心是其问题库,它包含了与《火影忍者》相关的各种问题。开发人员需要设计和构建这个知识库,并确保问题的多样性和覆盖面。
4. **逻辑和流程控制:** 在应用程序中,需要编写逻辑来控制测验的流程,比如问题的随机出现、计时器、计分机制以及结束时的反馈。
5. **用户界面(UI)交互:** 尽管是CLI,用户界面仍然重要。开发者需要确保用户体验流畅,这包括清晰的问题呈现、简洁的指令和友好的输出格式。
6. **模块化和封装:** 开发过程中应当遵循模块化原则,将不同的功能分隔开来,以便于管理和维护。例如,可以将问题生成器、计分器和用户输入处理器等封装成独立的模块。
7. **单元测试和调试:** 测验应用程序在发布前需要经过严格的测试和调试。使用如Mocha或Jest这样的JavaScript测试框架可以编写单元测试,并通过控制台输出调试信息来排除故障。
8. **部署和分发:** 最后,开发完成的应用程序需要被打包和分发。如果是基于Node.js的应用程序,常见的做法是将其打包为可执行文件(如使用electron或pkg工具),以便在不同的操作系统上运行。
根据提供的文件信息,虽然具体细节有限,但可以推测该应用程序可能采用了上述技术点。用户通过点击提供的链接,可能将被引导到一个网页或直接下载CLI应用程序的可执行文件,从而开始进行《火影忍者》的知识测验。通过这个测验,用户不仅能享受答题的乐趣,还可以加深对《火影忍者》的理解和认识。