rdkit怎么计算logp
时间: 2024-06-11 15:06:30 浏览: 207
RDKit是一种化学信息学工具包,可以用来计算化合物的物理化学性质。计算化合物的LogP(分配系数)需要进行以下步骤:
1. 读入分子结构:使用RDKit中的MolFromSmiles函数读入分子的SMILES字符串或者MolFromMolFile函数读入分子的mol文件。
2. 生成描述符:使用RDKit中的Descriptors模块计算描述符,其中包括荷电性、脂水平面面积、极性表面积等等。
3. 计算LogP:使用RDKit中的MolLogP函数计算分子的LogP值。
下面是一个使用RDKit计算LogP的示例代码:
```python
from rdkit import Chem
from rdkit.Chem import Descriptors
# 读入分子结构
smiles = 'CC(=O)Oc1ccccc1C(=O)O'
mol = Chem.MolFromSmiles(smiles)
# 生成描述符
mw = Descriptors.MolWt(mol)
logp = Descriptors.MolLogP(mol)
# 输出LogP值
print('LogP:', logp)
```
输出结果为:
```
LogP: 2.6724000000000003
```
以上代码将读入一个苯乙酸分子的SMILES字符串,使用Descriptors模块计算分子的描述符,然后使用MolLogP函数计算分子的LogP值。最后输出LogP值为2.67。
相关问题
rdkit 计算片段
RDKit 是一个非常强大的分子工具包,可以用来计算分子的各种性质,包括分子片段。
要计算分子的片段,可以使用 RDKit 中的 MolFragmentToSmiles() 函数,该函数可以将分子中的一个片段转换为 SMILES 字符串。例如,以下代码将计算苯环的 SMILES 字符串:
```python
from rdkit import Chem
mol = Chem.MolFromSmiles("c1ccccc1")
frag_smiles = Chem.MolFragmentToSmiles(mol, atomsToUse=range(6))
print(frag_smiles)
```
输出结果为:
```
c1ccccc1
```
其中 atomsToUse 参数指定了要使用的原子的索引,这里使用了范围为 0 到 5 的索引,即苯环的 6 个原子。
除了 MolFragmentToSmiles() 函数外,RDKit 还提供了许多其他的函数用于计算分子片段,例如 MolFragmentToSmarts() 函数可以将分子片段转换为 SMARTS 字符串。可以根据具体的需求选择适合的函数进行计算。
rdkit计算分子片段
RDKit提供了多种函数来计算分子片段,其中一种常用的函数是MolFragmentToSmiles。该函数可以将分子中的一个片段转换为SMILES字符串。
以下是一个计算分子片段的示例代码:
```python
from rdkit import Chem
mol = Chem.MolFromSmiles('COC1=CC=C(C=C1)C2=CC=C(C=C2)[N+](=O)[O-]')
submol = Chem.MolFromSmiles('COC')
smiles = Chem.MolFragmentToSmiles(mol, submol.GetSubstructMatch(mol))
print(smiles)
```
在上面的代码中,我们首先使用MolFromSmiles函数将SMILES字符串转换为分子对象,然后使用MolFromSmiles函数将要计算的分子片段转换为分子对象。接着,我们使用GetSubstructMatch函数找到分子中与分子片段匹配的子结构,最后使用MolFragmentToSmiles函数计算分子片段的SMILES字符串。
输出结果为:
```
COC
```
这表明输入分子中包含有一个与分子片段匹配的子结构,并且该子结构对应的SMILES字符串为COC。
相关推荐
最新推荐
面向多场景应用的光网络通感一体化架构和关键技术方案研究.pdf
面向多场景应用的光网络通感一体化架构和关键技术方案研究
基于Vue框架的Digital Twin开发设计源码
该项目是基于Vue框架的Digital Twin开发设计源码,由38个文件组成,涉及TypeScript、JavaScript、Vue、CSS、HTML等多种编程语言,包括9个TypeScript文件、6个JavaScript文件、5个JSON文件、5个Vue文件、3个SVG文件、2个Markdown文件、2个WebAssembly文件、1个Git忽略文件、1个HTML文件和1个CSS文件。该源码旨在提供大学生在线学术交流的平台,助力学术创新与协作。
IPQ4019 QSDK开源代码资源包发布
资源摘要信息:"IPQ4019是高通公司针对网络设备推出的一款高性能处理器,它是为需要处理大量网络流量的网络设备设计的,例如无线路由器和网络存储设备。IPQ4019搭载了强大的四核ARM架构处理器,并且集成了一系列网络加速器和硬件加密引擎,确保网络通信的速度和安全性。由于其高性能的硬件配置,IPQ4019经常用于制造高性能的无线路由器和企业级网络设备。
QSDK(Qualcomm Software Development Kit)是高通公司为了支持其IPQ系列芯片(包括IPQ4019)而提供的软件开发套件。QSDK为开发者提供了丰富的软件资源和开发文档,这使得开发者可以更容易地开发出性能优化、功能丰富的网络设备固件和应用软件。QSDK中包含了内核、驱动、协议栈以及用户空间的库文件和示例程序等,开发者可以基于这些资源进行二次开发,以满足不同客户的需求。
开源代码(Open Source Code)是指源代码可以被任何人查看、修改和分发的软件。开源代码通常发布在公共的代码托管平台,如GitHub、GitLab或SourceForge上,它们鼓励社区协作和知识共享。开源软件能够通过集体智慧的力量持续改进,并且为开发者提供了一个测试、验证和改进软件的机会。开源项目也有助于降低成本,因为企业或个人可以直接使用社区中的资源,而不必从头开始构建软件。
U-Boot是一种流行的开源启动加载程序,广泛用于嵌入式设备的引导过程。它支持多种处理器架构,包括ARM、MIPS、x86等,能够初始化硬件设备,建立内存空间的映射,从而加载操作系统。U-Boot通常作为设备启动的第一段代码运行,它为系统提供了灵活的接口以加载操作系统内核和文件系统。
标题中提到的"uci-2015-08-27.1.tar.gz"是一个开源项目的压缩包文件,其中"uci"很可能是指一个具体项目的名称,比如U-Boot的某个版本或者是与U-Boot配置相关的某个工具(U-Boot Config Interface)。日期"2015-08-27.1"表明这是该项目的2015年8月27日的第一次更新版本。".tar.gz"是Linux系统中常用的归档文件格式,用于将多个文件打包并进行压缩,方便下载和分发。"
描述中复述了标题的内容,强调了文件是关于IPQ4019处理器的QSDK资源,且这是一个开源代码包。此处未提供额外信息。
标签"软件/插件"指出了这个资源的性质,即它是一个软件资源,可能包含程序代码、库文件或者其他可以作为软件一部分的插件。
在文件名称列表中,"uci-2015-08-27.1"与标题保持一致,表明这是一个特定版本的软件或代码包。由于实际的文件列表中只提供了这一项,我们无法得知更多的文件信息,但可以推测这是一个单一文件的压缩包。
管理建模和仿真的文件
管理Boualem Benatallah引用此版本:布阿利姆·贝纳塔拉。管理建模和仿真。约瑟夫-傅立叶大学-格勒诺布尔第一大学,1996年。法语。NNT:电话:00345357HAL ID:电话:00345357https://theses.hal.science/tel-003453572008年12月9日提交HAL是一个多学科的开放存取档案馆,用于存放和传播科学研究论文,无论它们是否被公开。论文可以来自法国或国外的教学和研究机构,也可以来自公共或私人研究中心。L’archive ouverte pluridisciplinaire
【PDF表格创建与编辑】:使用Apache PDFBox和iText库制作表格,表格处理不求人
# 1. PDF表格创建与编辑概述
在当今数字化时代,PDF格式因其可移植性和可维护性成为了电子文档的首选。在诸多应用场景中,表格作为一种重要的信息传递工具,其创建与编辑的需求日益增长。本章节将为读者提供一个关于PDF表格创建与编辑的概述,为后续章节中使用Apache PDFBox和iText库进行深入探讨奠定基础。
## 1.1 PDF表格的作用与重要性
PDF表格在商务沟通、数据分析
python输出txt文件
在Python中,你可以使用内置的`open()`函数来创建和写入TXT文件。下面是一个简单的例子:
```python
# 打开文件(如果不存在则创建)
with open('example.txt', 'w') as file:
# 写入文本内容
file.write('这是你要写入的内容')
# 如果你想追加内容而不是覆盖原有文件
# 使用 'a' 模式(append)
# with open('example.txt', 'a') as file:
# file.write('\n这是追加的内容')
# 关闭文件时会自动调用 `close()` 方法,但使
高频组电赛必备:掌握数字频率合成模块要点
资源摘要信息:"2022年电赛 高频组必备模块 数字频率合成模块"
数字频率合成(DDS,Direct Digital Synthesis)技术是现代电子工程中的一种关键技术,它允许通过数字方式直接生成频率可调的模拟信号。本模块是高频组电赛参赛者必备的组件之一,对于参赛者而言,理解并掌握其工作原理及应用是至关重要的。
本数字频率合成模块具有以下几个关键性能参数:
1. 供电电压:模块支持±5V和±12V两种供电模式,这为用户提供了灵活的供电选择。
2. 外部晶振:模块自带两路输出频率为125MHz的外部晶振,为频率合成提供了高稳定性的基准时钟。
3. 输出信号:模块能够输出两路频率可调的正弦波信号。其中,至少有一路信号的幅度可以编程控制,这为信号的调整和应用提供了更大的灵活性。
4. 频率分辨率:模块提供的频率分辨率为0.0291Hz,这样的精度意味着可以实现非常精细的频率调节,以满足高频应用中的严格要求。
5. 频率计算公式:模块输出的正弦波信号频率表达式为 fout=(K/2^32)×CLKIN,其中K为设置的频率控制字,CLKIN是外部晶振的频率。这一计算方式表明了频率输出是通过编程控制的频率控制字来设定,从而实现高精度的频率合成。
在高频组电赛中,参赛者不仅需要了解数字频率合成模块的基本特性,还应该能够将这一模块与其他模块如移相网络模块、调幅调频模块、AD9854模块和宽带放大器模块等结合,以构建出性能更优的高频信号处理系统。
例如,移相网络模块可以实现对信号相位的精确控制,调幅调频模块则能够对信号的幅度和频率进行调整。AD9854模块是一种高性能的DDS芯片,可以用于生成复杂的波形。而宽带放大器模块则能够提供足够的增益和带宽,以保证信号在高频传输中的稳定性和强度。
在实际应用中,电赛参赛者需要根据项目的具体要求来选择合适的模块组合,并进行硬件的搭建与软件的编程。对于数字频率合成模块而言,还需要编写相应的控制代码以实现对K值的设定,进而调节输出信号的频率。
交流与讨论在电赛准备过程中是非常重要的。与队友、指导老师以及来自同一领域的其他参赛者进行交流,不仅可以帮助解决技术难题,还可以相互启发,激发出更多创新的想法和解决方案。
总而言之,对于高频组的电赛参赛者来说,数字频率合成模块是核心组件之一。通过深入了解和应用该模块的特性,结合其他模块的协同工作,参赛者将能够构建出性能卓越的高频信号处理设备,从而在比赛中取得优异成绩。
"互动学习:行动中的多样性与论文攻读经历"
多样性她- 事实上SCI NCES你的时间表ECOLEDO C Tora SC和NCESPOUR l’Ingén学习互动,互动学习以行动为中心的强化学习学会互动,互动学习,以行动为中心的强化学习计算机科学博士论文于2021年9月28日在Villeneuve d'Asq公开支持马修·瑟林评审团主席法布里斯·勒菲弗尔阿维尼翁大学教授论文指导奥利维尔·皮耶昆谷歌研究教授:智囊团论文联合主任菲利普·普雷教授,大学。里尔/CRISTAL/因里亚报告员奥利维耶·西格德索邦大学报告员卢多维奇·德诺耶教授,Facebook /索邦大学审查员越南圣迈IMT Atlantic高级讲师邀请弗洛里安·斯特鲁布博士,Deepmind对于那些及时看到自己错误的人...3谢谢你首先,我要感谢我的两位博士生导师Olivier和Philippe。奥利维尔,"站在巨人的肩膀上"这句话对你来说完全有意义了。从科学上讲,你知道在这篇论文的(许多)错误中,你是我可以依
【PDF元数据管理】:如何使用Java库管理和编辑PDF元数据,元数据管理的秘密
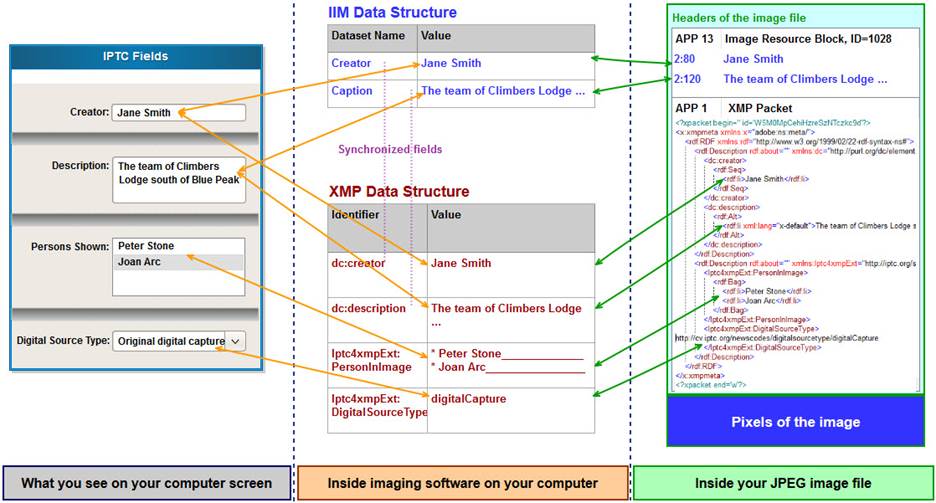
# 1. PDF元数据管理概述
在当今数字化工作流程中,PDF文件以其平台独立性和格式固定性成为了文档交换的标准格式。元数据——关于数据的数据——在PDF中扮演了至关重要的角色,它们提供了文档的内容摘要和结构信息,使得文件管理更加高效。在本章中,我们将探讨PDF元数据的基础知识,它们如何增强文档的可用性,
TypeScript 二进制16在代码里怎么表示
在TypeScript中,如果你想表示二进制数16,你可以使用字符串字面量或者专门的二进制格式来表示。这里有几种常见的方法:
1. **字符串字面量**:
```typescript
let binaryNumber: string = "0x10"; // 使用 '0x' 前缀标识这是一个十六进制数
```
或者直接写成:
```typescript
let binaryNumber: string = "10"; // 这种形式适用于0-9的二进制数,TypeScript会自动识别其为十进制转义序列
```
2. **Number类型的数值**:
如果你想要将其转换为Number类型,