# 初始化变量 output = [['id'] + list(range(1, len(lines[1].strip()) + 1))] current_id = '' current_seq = [] # 循环遍历所有行 for line in lines: # 如果是一个新的序列的id行 if line.startswith('>'): # 如果之前已经读取了一个序列的id和序列 if current_id != '': # 将当前的id和序列添加到输出列表中 output.append([current_id] + current_seq) # 重置当前id和序列 current_id = '' current_seq = [] # 获取新的序列的id current_id = line.strip()[1:] # 如果是一个序列行 else: # 将碱基一个一个地添加到列表中 nucleotides = list(line.strip()) nucleotides_with_spacer = [nucleotide + ' ' for nucleotide in nucleotides] current_seq.append(''.join(nucleotides_with_spacer)) # 将最后一个序列添加到输出列表中 output.append([current_id] + current_seq) # 将输出列表写入到csv文件中 with open('output.csv', 'w', newline='') as f: writer = csv.writer(f, delimiter=',', quoting=csv.QUOTE_NONE) writer.writerows(output)
时间: 2024-04-01 13:36:07 浏览: 17
这段代码是一个 Python 代码片段,它主要是将一个多行的 DNA 序列文件(FASTA 格式)转换为 CSV 文件格式。代码执行的过程如下:
1. 初始化输出变量 `output`,其中包含一个表头行,表头行的第一列是 `'id'`,其余列是从 1 开始递增的数字。
2. 初始化变量 `current_id` 和 `current_seq`,分别用于存储当前正在处理的 DNA 序列的 ID 和序列。
3. 循环遍历输入文件中的所有行。
4. 如果当前行以 `>` 开头,则表示当前行是一个新的 DNA 序列的 ID 行。
5. 如果当前行不是 ID 行,则表示当前行是一个 DNA 序列的碱基行,将碱基一个一个地添加到 `current_seq` 列表中。
6. 将处理完成的 DNA 序列的 ID 和序列添加到输出列表 `output` 中。
7. 将输出列表写入到 CSV 文件中。
该代码还包含了一些细节处理,例如将每个碱基后面加上一个空格,以便在 CSV 文件中更好地显示。
相关问题
import csv # 打开fasta文件 with open('your_fasta_file.fasta', 'r') as f: # 读取所有行 lines = f.readlines() # 初始化变量 output = [['id'] + list(range(1, len(lines[1].strip()) + 1))] current_id = '' current_seq = '' # 循环遍历所有行 for line in lines: # 如果是一个新的序列的id行 if line.startswith('>'): # 如果之前已经读取了一个序列的id和序列 if current_id != '': # 将当前的id和序列添加到输出列表中 output.append([current_id] + current_seq) # 重置当前id和序列 current_id = '' current_seq = [] # 获取新的序列的id current_id = line.strip()[1:] # 如果是一个序列行 else: # 将碱基添加到当前序列中 current_seq += list(line.strip()) # 将最后一个序列添加到输出列表中 output.append([current_id] + current_seq) # 将输出列表写入到csv文件中 with open('output.csv', 'w', newline='') as f: writer = csv.writer(f) writer.writerows(output)这个报错can only concatenate str (not "list") to str
这个错误是因为在如下代码中,你将列表 `current_seq` 和字符串进行了拼接:
```
output.append([current_id] + current_seq)
```
这里的 `[current_id]` 是一个只包含一个字符串元素的列表,而 `current_seq` 是一个列表,两者进行拼接时会报错。
要解决这个问题,你需要将 `current_seq` 列表转换为一个字符串,然后再将其与 `[current_id]` 列表进行拼接。可以使用 join() 方法将列表转换为字符串,示例代码如下:
```
output.append([current_id] + [''.join(current_seq)])
```
这段代码将列表 `current_seq` 转换为字符串,然后将其与列表 `[current_id]` 进行拼接,输出结果为一个包含 id 和序列的列表。
import csv # 打开fasta文件 with open('E:\泛基因组分析\ORF\ORF_HMMER_extrct\ORF_FAS\ORF4.fasta', 'r') as f: # 读取所有行 lines = f.readlines() # 初始化变量 output = [['id'] + list(range(1, len(lines[1].strip()) + 1))] current_id = '' current_seq = [] # 循环遍历所有行 for line in lines: # 如果是一个新的序列的id行 if line.startswith('>'): # 如果之前已经读取了一个序列的id和序列 if current_id != '': # 将当前的id和序列添加到输出列表中 output.append([current_id] + current_seq) # 重置当前id和序列 current_id = '' current_seq = [] # 获取新的序列的id current_id = line.strip()[1:] # 如果是一个序列行 else: # 将碱基添加到当前序列中 nucleotides = list(line.strip()) nucleotides_with_spacer = ', '.join(nucleotides) current_seq.append(nucleotides_with_spacer) # 将最后一个序列添加到输出列表中 output.append([current_id] + [''] + current_seq[:-1] + [current_seq[-1].replace(',','')]) # 在最后添加一个空行 output.append([]) # 将输出列表写入到csv文件中 with open('E:\泛基因组分析\ORF\ORF_HMMER_extrct\ORF_FAS\output.csv', 'w', newline='') as f: writer = csv.writer(f, delimiter='\t', quoting=csv.QUOTE_NONE) writer.writerows(output)
这段代码的作用是将一个 fasta 格式的文件转换为 csv 格式的文件,并将每个序列的碱基序列转换为以逗号和空格分隔的字符串,每行写入到 csv 文件中。其中,每个序列的 id 作为 csv 文件的一列,碱基序列作为 csv 文件的另一列。
如果您想在生成的 csv 文件中添加回车符,您可以在写入 csv 文件的时候手动添加回车符。您可以在每行的末尾添加一个回车符和换行符(`\r\n`),确保每行都能够被分隔开。例如:
```python
import csv
# 打开fasta文件
with open('E:\泛基因组分析\ORF\ORF_HMMER_extrct\ORF_FAS\ORF4.fasta', 'r') as f:
# 读取所有行
lines = f.readlines()
# 初始化变量
output = [['id'] + list(range(1, len(lines[1].strip()) + 1))]
current_id = ''
current_seq = []
# 循环遍历所有行
for line in lines:
# 如果是一个新的序列的id行
if line.startswith('>'):
# 如果之前已经读取了一个序列的id和序列
if current_id != '':
# 将当前的id和序列添加到输出列表中
output.append([current_id] + [''] + current_seq[:-1] + [current_seq[-1].replace(',', '')])
# 在当前序列的碱基序列中添加回车符
current_seq = [', '.join(nucleotides) + '\r\n' for nucleotides in current_seq]
# 获取新的序列的id
current_id = line.strip()[1:]
# 如果是一个序列行
else:
# 将碱基添加到当前序列中
nucleotides = list(line.strip())
current_seq.append(nucleotides)
# 将最后一个序列添加到输出列表中
output.append([current_id] + [''] + current_seq[:-1] + [current_seq[-1].replace(',', '')])
# 在最后添加一个空行
output.append([])
# 将输出列表写入到csv文件中
with open('E:\泛基因组分析\ORF\ORF_HMMER_extrct\ORF_FAS\output.csv', 'w', newline='') as f:
writer = csv.writer(f, delimiter='\t', quoting=csv.QUOTE_NONE)
for row in output:
writer.writerow(row)
f.write('\r\n')
```
在这个例子中,我们在写入每一行的时候手动添加了回车符和换行符,确保了每行都能够被分隔开。如果您有其他问题,请随时提出,我会尽力帮助您。
相关推荐
最新推荐
C++全局变量初始化的一点总结
注意:本文所说的全局变量指的是 variables with static storage,措词来自 c++ 的语言标准文档。 什么时候初始化 ... static initialization: 静态初始化指的是用常量来对变量进行初始化,主要包括 zero init
MDK下怎样才能让变量在复位时不被初始化
最近一个项目需要保存一下临时数据,而产品容易受干扰而...所以需要保存一下数据,那么只有在系统复位时候不再初始化变量即可。对应MDK(keil)来说是一个比较麻烦的问题。通过网络上找了大量资料和测试发现终于可以了。
Java中static静态变量的初始化完全解析
static所声明的变量在Java中有一个初始化的先后顺序,带着这个问题接下来我们就来进行Java中static静态变量的初始化完全解析:
node-v18.18.2-headers.tar.xz
Node.js,简称Node,是一个开源且跨平台的JavaScript运行时环境,它允许在浏览器外运行JavaScript代码。Node.js于2009年由Ryan Dahl创立,旨在创建高性能的Web服务器和网络应用程序。它基于Google Chrome的V8 JavaScript引擎,可以在Windows、Linux、Unix、Mac OS X等操作系统上运行。
Node.js的特点之一是事件驱动和非阻塞I/O模型,这使得它非常适合处理大量并发连接,从而在构建实时应用程序如在线游戏、聊天应用以及实时通讯服务时表现卓越。此外,Node.js使用了模块化的架构,通过npm(Node package manager,Node包管理器),社区成员可以共享和复用代码,极大地促进了Node.js生态系统的发展和扩张。
Node.js不仅用于服务器端开发。随着技术的发展,它也被用于构建工具链、开发桌面应用程序、物联网设备等。Node.js能够处理文件系统、操作数据库、处理网络请求等,因此,开发者可以用JavaScript编写全栈应用程序,这一点大大提高了开发效率和便捷性。
在实践中,许多大型企业和组织已经采用Node.js作为其Web应用程序的开发平台,如Netflix、PayPal和Walmart等。它们利用Node.js提高了应用性能,简化了开发流程,并且能更快地响应市场需求。
node-v7.7.3-headers.tar.xz
Node.js,简称Node,是一个开源且跨平台的JavaScript运行时环境,它允许在浏览器外运行JavaScript代码。Node.js于2009年由Ryan Dahl创立,旨在创建高性能的Web服务器和网络应用程序。它基于Google Chrome的V8 JavaScript引擎,可以在Windows、Linux、Unix、Mac OS X等操作系统上运行。
Node.js的特点之一是事件驱动和非阻塞I/O模型,这使得它非常适合处理大量并发连接,从而在构建实时应用程序如在线游戏、聊天应用以及实时通讯服务时表现卓越。此外,Node.js使用了模块化的架构,通过npm(Node package manager,Node包管理器),社区成员可以共享和复用代码,极大地促进了Node.js生态系统的发展和扩张。
Node.js不仅用于服务器端开发。随着技术的发展,它也被用于构建工具链、开发桌面应用程序、物联网设备等。Node.js能够处理文件系统、操作数据库、处理网络请求等,因此,开发者可以用JavaScript编写全栈应用程序,这一点大大提高了开发效率和便捷性。
在实践中,许多大型企业和组织已经采用Node.js作为其Web应用程序的开发平台,如Netflix、PayPal和Walmart等。它们利用Node.js提高了应用性能,简化了开发流程,并且能更快地响应市场需求。
zigbee-cluster-library-specification
最新的zigbee-cluster-library-specification说明文档。
管理建模和仿真的文件
管理Boualem Benatallah引用此版本:布阿利姆·贝纳塔拉。管理建模和仿真。约瑟夫-傅立叶大学-格勒诺布尔第一大学,1996年。法语。NNT:电话:00345357HAL ID:电话:00345357https://theses.hal.science/tel-003453572008年12月9日提交HAL是一个多学科的开放存取档案馆,用于存放和传播科学研究论文,无论它们是否被公开。论文可以来自法国或国外的教学和研究机构,也可以来自公共或私人研究中心。L’archive ouverte pluridisciplinaire
实现实时数据湖架构:Kafka与Hive集成
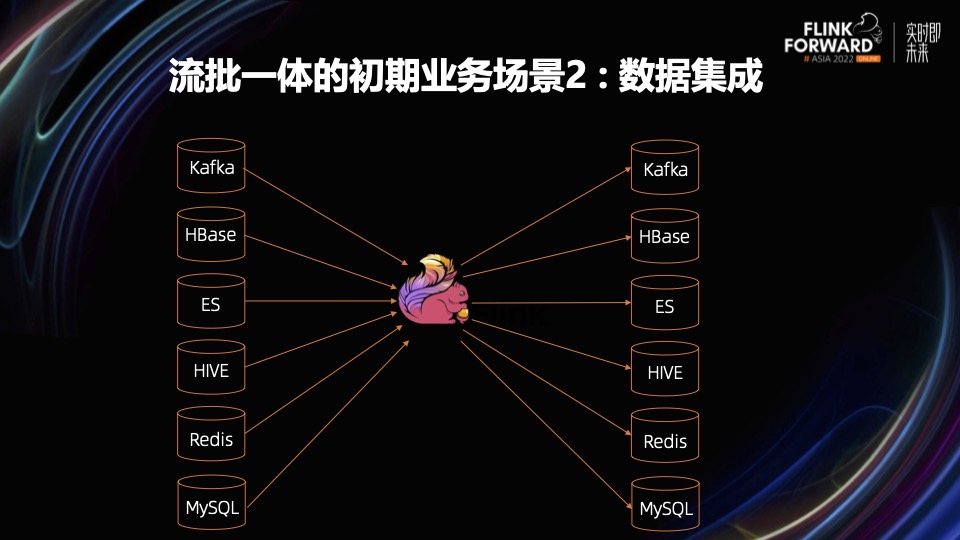
# 1. 实时数据湖架构概述**
实时数据湖是一种现代数据管理架构,它允许企业以低延迟的方式收集、存储和处理大量数据。与传统数据仓库不同,实时数据湖不依赖于预先定义的模式,而是采用灵活的架构,可以处理各种数据类型和格式。这种架构为企业提供了以下优势:
- **实时洞察:**实时数据湖允许企业访问最新的数据,从而做出更明智的决策。
- **数据民主化:**实时数据湖使各种利益相关者都可
可见光定位LED及其供电硬件具体型号,广角镜头和探测器,实验设计具体流程步骤,
1. 可见光定位LED型号:一般可使用5mm或3mm的普通白色LED,也可以选择专门用于定位的LED,例如OSRAM公司的SFH 4715AS或Vishay公司的VLMU3500-385-120。
2. 供电硬件型号:可以使用常见的直流电源供电,也可以选择专门的LED驱动器,例如Meanwell公司的ELG-75-C或ELG-150-C系列。
3. 广角镜头和探测器型号:一般可采用广角透镜和CMOS摄像头或光电二极管探测器,例如Omron公司的B5W-LA或Murata公司的IRS-B210ST01。
4. 实验设计流程步骤:
1)确定实验目的和研究对象,例如车辆或机器人的定位和导航。
JSBSim Reference Manual
JSBSim参考手册,其中包含JSBSim简介,JSBSim配置文件xml的编写语法,编程手册以及一些应用实例等。其中有部分内容还没有写完,估计有生之年很难看到完整版了,但是内容还是很有参考价值的。