通过网络查询连接函数使用方法,当关键字列名不一样时,如何代码实现? 例如: credit_t <- tibble(课程名 = colnames(score_t)[-1]) |> left_join(credit, by = _________) credit_t
时间: 2023-05-17 16:07:05 浏览: 249
可以使用参数 `by.x` 和 `by.y` 来指定连接的列名不一样时的对应关系,具体代码实现如下:
credit_t <- tibble(课程名 = colnames(score_t)[-1]) |>
left_join(credit, by = c("课程名" = "course_name"))
相关问题
运行下列代码,将基于 stringr:: words 的数据生成一个tibble,命名为tb_wordstb_words <- tibble( index = seq_along(stringr::words), words = stringr::words)
好的,可以运行以下代码将 `stringr::words` 生成的数据转化为 `tibble` 格式,并命名为 `tb_words`:
```R
library(tibble)
library(stringr)
tb_words <- tibble(
index = seq_along(stringr::words),
words = stringr::words
)
```
这里使用了 `seq_along()` 函数来生成一个整数序列,作为表格的第一列 `index`。同时,使用 `stringr::words` 函数生成单词向量,并将其作为表格的第二列 `words`。最后将这两列合并成一个 `tibble`,命名为 `tb_words`。
PCA_Plot_3=function (data,Annotation,VAR,Color) { # logcountdata row:genes,column: samples pca <- prcomp(data) pca_out<-as.data.frame(pca$x) df_out<- pca_out %>%tibble::rownames_to_column(var=VAR) %>% left_join(., Annotation) #df_out<- merge (pca_out,Annotation,by.x=0,by.y=0) # label_color<- factor(df_out[,group]) ggplot(df_out,aes_string(x="PC1",y="PC2")) +geom_point(aes_string(colour = Color)) } Deseq2_Deseq_function_2=function (Countdata,Coldata) { dds_fil <- DESeq2:: DESeqDataSetFromMatrix(countData =Countdata, colData = Coldata, design = ~Group) dds_fil_Deg<- DESeq2::DESeq(dds_fil) return(dds_fil_Deg) } pheatmap_singscore=function (pathways,data,Annotation) { Gene_select_anno= data[,colnames(data) %in% pathways] %>%t()%>%.[,rownames(Annotation)] # return(Gene_select_anno) # Anno_expression_data=Gene_select_anno[,c("SYMBOL",Group_select)] %>% as.data.frame() %>% distinct() %>% na.omit() # rownames(Anno_expression_data)=Anno_expression_data[,"SYMBOL"] # Annotation=group_anno["Gene_type"] # input= Anno_expression_data[,Group_select] # F2_pheatmap <- pheatmap::pheatmap(input, cellwigermline calling GATKdth = 10, cellheight = 12, scale = "row", # treeheight_row = 5, # show_rownames = T,show_colnames = T, # annotation_col= Annotation, # # annotation_row=Annotation, # annotation_legend=Label_def, # cluster_rows = T, cluster_cols = F,clustering_distance_rows = "euclidean") pheatmap::pheatmap(Gene_select_anno, cellwigermline=5, cellheight = 10,cellwidth = 10, scale = "row", treeheight_row = 5, show_rownames = T,show_colnames = F, annotation_col= Annotation, # annotation_row=Annotation, #annotation_legend=Label_def, cluster_rows = T, cluster_cols = F,clustering_distance_rows = "euclidean") } matrix.please<-function(x) { m<-as.matrix(x[,-1]) rownames(m)<-x[,1] m } 这是r语言的代码,告诉我每一条代码的作用和意义
PCA_Plot_3: 这个函数用来绘制主成分分析(PCA)的散点图。它接受四个参数:data(数据矩阵),Annotation(注释信息),VAR(行名),Color(颜色)。首先,它对数据进行主成分分析(prcomp函数),然后将主成分分析的结果转换为数据帧(pca_out)。接下来,它将行名转换为指定的VAR变量名,并将注释信息与主成分分析结果进行连接(left_join函数)。最后,它使用ggplot库绘制散点图,其中x轴表示PC1,y轴表示PC2,点的颜色由Color参数指定。
Deseq2_Deseq_function_2: 这个函数用于进行差异表达分析。它接受两个参数:Countdata(基因计数数据)和Coldata(样本注释信息)。首先,它使用DESeq2包中的DESeqDataSetFromMatrix函数将Countdata和Coldata转换为差异表达分析所需的数据集对象。然后,它使用DESeq2包中的DESeq函数进行差异表达分析,并返回结果。
pheatmap_singscore: 这个函数用于绘制热图。它接受三个参数:pathways(基因通路信息),data(基因表达数据),Annotation(注释信息)。首先,它从data中选择pathways对应的基因,并根据Annotation的行名对选定的基因进行筛选。然后,它使用pheatmap包中的pheatmap函数绘制热图,其中Gene_select_anno是待绘制的基因数据,Annotation用于注释列,cluster_rows参数表示是否对行进行聚类,clustering_distance_rows参数表示行聚类所使用的距离度量。
matrix.please: 这个函数用于将数据框转换为矩阵。它接受一个参数x(数据框),并将x的第一列作为行名,将x的其余列转换为矩阵。最后,它返回该矩阵。
阅读全文
相关推荐
大家在看
二阶有源带通滤波器设计及参数计算.doc
二阶有源带通滤波器设计及参数计算,有详细的计算过程,应用在全国大学生电子设计竞赛的综合测评里,提取基波,三次谐波,五次谐波
基于Android Studio开发的安卓的通讯录管理app
功能包含:新增联系人、编辑联系人、删除联系人、拨打电话、发送短信等相关操作。 资源包含源码:1、apk安装包 2、演示视频 3、 基本安装环境、4、运行文档 5、以及源代码
seadas海洋遥感软件使用说明
这是一个海洋遥感软件seadas的使用文档,希望这个资料能对学习海洋遥感的朋友有所帮助
DX200 使用說明書.pdf
安川機器人DX200系統控制文檔,包含安川机器人操作说明,安裝解說,基础操作,使用和簡易设置说明。
[ExDui自绘]动态创建控件-易语言
功能简单,动态创建控件 排列
方便自己使用的一个小工具
(图中数据仅测试用而已)
最新推荐
科研工作量管理系统(代码+数据库+LW)
摘 要
现代经济快节奏发展以及不断完善升级的信息化技术,让传统数据信息的管理升级为软件存储,归纳,集中处理数据信息的管理方式。本科研工作量管理系统就是在这样的大环境下诞生,其可以帮助管理者在短时间内处理完毕庞大的数据信息,使用这种软件工具可以帮助管理人员提高事务处理效率,达到事半功倍的效果。此科研工作量管理系统利用当下成熟完善的SSM框架,使用跨平台的可开发大型商业网站的Java语言,以及最受欢迎的RDBMS应用软件之一的Mysql数据库进行程序开发。实现了用户在线选择试题并完成答题,在线查看考核分数。管理员管理字典管理、工作量管理、科研获奖管理、科研论文管理、秘书管理、科研项目管理、教师管理、管理员管理等功能。科研工作量管理系统的开发根据操作人员需要设计的界面简洁美观,在功能模块布局上跟同类型网站保持一致,程序在实现基本要求功能时,也为数据信息面临的安全问题提供了一些实用的解决方案。可以说该程序在帮助管理者高效率地处理工作事务的同时,也实现了数据信息的整体化,规范化与自动化。
关键词:科研工作量管理系统;SSM框架;Mysql;自动化
基于遗产算法的多目标分布式电源选址定容 以投资成本、网络损耗和系统电压稳定性为目标实现分布式电源选址定容,通过IEEE33节点系统进行仿真验证,结果如下图所示
基于遗产算法的多目标分布式电源选址定容
以投资成本、网络损耗和系统电压稳定性为目标实现分布式电源选址定容,通过IEEE33节点系统进行仿真验证,结果如下图所示
jh_flutter_demo.apk
jh_flutter_demo.apk
windows jdk 8 ,jdk 11, jdk 17
windows jdk 8 ,jdk 11, jdk 17
租赁合同编写指南及下载资源
资源摘要信息:《租赁合同》是用于明确出租方与承租方之间的权利和义务关系的法律文件。在实际操作中,一份详尽的租赁合同对于保障交易双方的权益至关重要。租赁合同应当包括但不限于以下要点:
1. 双方基本信息:租赁合同中应明确出租方(房东)和承租方(租客)的名称、地址、联系方式等基本信息。这对于日后可能出现的联系、通知或法律诉讼具有重要意义。
2. 房屋信息:合同中需要详细说明所租赁的房屋的具体信息,包括房屋的位置、面积、结构、用途、设备和家具清单等。这些信息有助于双方对租赁物有清晰的认识。
3. 租赁期限:合同应明确租赁开始和结束的日期,以及租期的长短。租赁期限的约定关系到租金的支付和合同的终止条件。
4. 租金和押金:租金条款应包括租金金额、支付周期、支付方式及押金的数额。同时,应明确规定逾期支付租金的处理方式,以及押金的退还条件和时间。
5. 维修与保养:在租赁期间,房屋的维护和保养责任应明确划分。通常情况下,房东负责房屋的结构和主要设施维修,而租客需负责日常维护及保持房屋的清洁。
6. 使用与限制:合同应规定承租方可以如何使用房屋以及可能的限制。例如,禁止非法用途、允许或禁止宠物、是否可以转租等。
7. 终止与续租:租赁合同应包括租赁关系的解除条件,如提前通知时间、违约责任等。同时,双方可以在合同中约定是否可以续租,以及续租的条件。
8. 解决争议的条款:合同中应明确解决可能出现的争议的途径,包括适用法律、管辖法院等,有助于日后纠纷的快速解决。
9. 其他可能需要的条款:根据具体情况,合同中可能还需要包括关于房屋保险、税费承担、合同变更等内容。
下载资源链接:【下载自www.glzy8.com管理资源吧】Rental contract.DOC
该资源为一份租赁合同模板,对需要进行房屋租赁的个人或机构提供了参考价值。通过对合同条款的详细列举和解释,该文档有助于用户了解和制定自己的租赁合同,从而在房屋租赁交易中更好地保护自己的权益。感兴趣的用户可以通过提供的链接下载文档以获得更深入的了解和实际操作指导。
【项目管理精英必备】:信息系统项目管理师教程习题深度解析(第四版官方教材全面攻略)
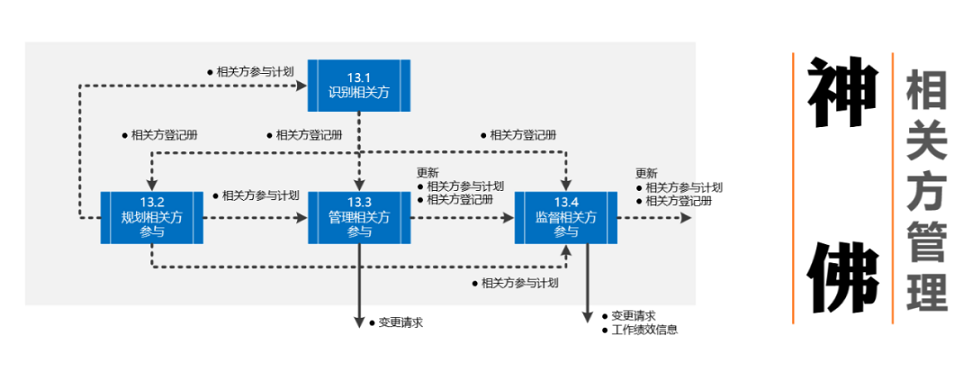
# 摘要
信息系统项目管理是确保项目成功交付的关键活动,涉及一系列管理过程和知识领域。本文深入探讨了信息系统项目管理的各个方面,包括项目管理过程组、知识领域、实践案例、管理工具与技术,以及沟通和团队协作。通过分析不同的项目管理方法论(如瀑布、迭代、敏捷和混合模型),并结合具体案例,文章阐述了项目管理的最佳实践和策略。此外,本文还涵盖了项目管理中的沟通管理、团队协作的重要性,
最具代表性的改进过的UNet有哪些?
UNet是一种广泛用于图像分割任务的卷积神经网络结构,它的特点是结合了下采样(编码器部分)和上采样(解码器部分),能够保留细节并生成精确的边界。为了提高性能和适应特定领域的需求,研究者们对原始UNet做了许多改进,以下是几个最具代表性的变种:
1. **DeepLab**系列:由Google开发,通过引入空洞卷积(Atrous Convolution)、全局平均池化(Global Average Pooling)等技术,显著提升了分辨率并保持了特征的多样性。
2. **SegNet**:采用反向传播的方式生成全尺寸的预测图,通过上下采样过程实现了高效的像素级定位。
3. **U-Net+
惠普P1020Plus驱动下载:办公打印新选择
资源摘要信息: "最新惠普P1020Plus官方驱动"
1. 惠普 LaserJet P1020 Plus 激光打印机概述:
惠普 LaserJet P1020 Plus 是惠普公司针对家庭、个人办公以及小型办公室(SOHO)市场推出的一款激光打印机。这款打印机的设计注重小巧体积和便携操作,适合空间有限的工作环境。其紧凑的设计和高效率的打印性能使其成为小型企业或个人用户的理想选择。
2. 技术特点与性能:
- 预热技术:惠普 LaserJet P1020 Plus 使用了0秒预热技术,能够极大减少打印第一张页面所需的等待时间,首页输出时间不到10秒。
- 打印速度:该打印机的打印速度为每分钟14页,适合处理中等规模的打印任务。
- 月打印负荷:月打印负荷高达5000页,保证了在高打印需求下依然能稳定工作。
- 标配硒鼓:标配的2000页打印硒鼓能够为用户提供较长的使用周期,减少了更换耗材的频率,节约了长期使用成本。
3. 系统兼容性:
驱动程序支持的操作系统包括 Windows Vista 64位版本。用户在使用前需要确保自己的操作系统版本与驱动程序兼容,以保证打印机的正常工作。
4. 市场表现:
惠普 LaserJet P1020 Plus 在上市之初便获得了市场的广泛认可,创下了百万销量的辉煌成绩,这在一定程度上证明了其可靠性和用户对其性能的满意。
5. 驱动程序文件信息:
压缩包内包含了适用于该打印机的官方驱动程序文件 "lj1018_1020_1022-HB-pnp-win64-sc.exe"。该文件是安装打印机驱动的执行程序,用户需要下载并运行该程序来安装驱动。
另一个文件 "jb51.net.txt" 从命名上来看可能是一个文本文件,通常这类文件包含了关于驱动程序的安装说明、版本信息或是版权信息等。由于具体内容未提供,无法确定确切的信息。
6. 使用场景:
由于惠普 LaserJet P1020 Plus 的打印速度和负荷能力,它适合那些需要快速、频繁打印文档的用户,例如行政助理、会计或小型法律事务所。它的紧凑设计也使得这款打印机非常适合在桌面上使用,从而不占用过多的办公空间。
7. 后续支持与维护:
用户在购买后可以通过惠普官方网站获取最新的打印机驱动更新以及技术支持。在安装新驱动之前,建议用户先卸载旧的驱动程序,以避免版本冲突或不必要的错误。
8. 其它注意事项:
- 用户在使用打印机时应注意按照官方提供的维护说明定期进行清洁和保养,以确保打印质量和打印机的使用寿命。
- 如果在打印过程中遇到任何问题,应先检查打印机设置、驱动程序是否正确安装以及是否有足够的打印纸张和墨粉。
综上所述,惠普 LaserJet P1020 Plus 是一款性能可靠、易于使用的激光打印机,特别适合小型企业或个人用户。正确的安装和维护可以确保其稳定和高效的打印能力,满足日常办公需求。
数字电路实验技巧:10大策略,让你的实验效率倍增!
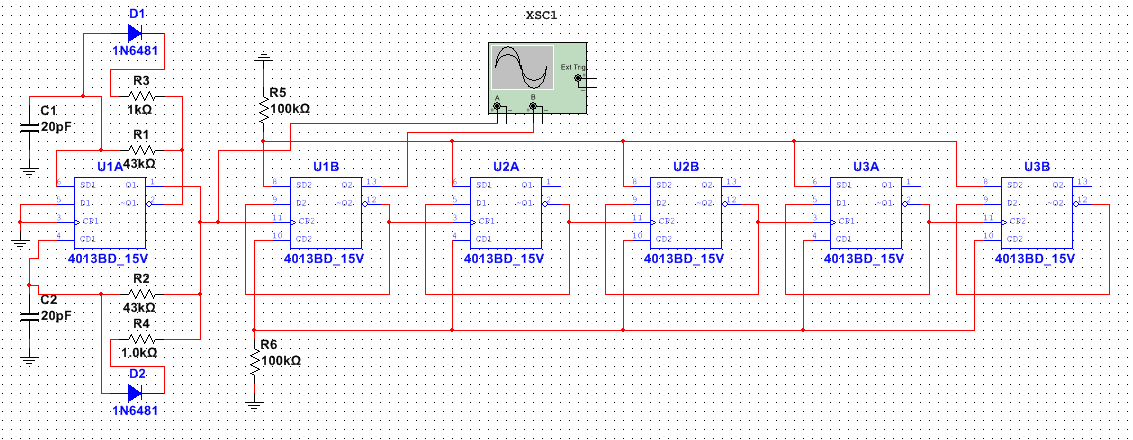
# 摘要
本论文详细介绍了数字电路实验的基础理论、设备使用、设计原则、实践操作、调试与故障排除以及报告撰写与成果展示。首先探讨了数字电路实验所需的基本理论和实验设备的种类与使用技巧,包括测量和故障诊断方法。接着,深入分析了电路设计的原则,涵盖设计流程、逻辑简化、优化策略及实验方案的制定。在实践操作章节中,具体
altium designer布线
### Altium Designer 布线教程和技巧
#### 一、环境设置与准备
为了更高效地完成布线工作,前期的准备工作至关重要。确保原理图已经完全无误并编译成功[^2]。
#### 二、同步查看原理图与PCB布局
通过在原理图标题栏处右键点击并选择 "Split Vertical" 可实现原理图和PCB视图的同时展示,这有助于理解电路连接关系以及提高布线效率。
#### 三、自动布线器配置
Altium Designer内置有强大的自动布线功能。进入“Tools -> PCB Rules and Constraints Editor”,可以自定义诸如最小间距、过孔尺寸等参数来满足