如何利用DFT计算优化石墨烯-偶氮苯分子复合物的光致异构化和热稳定性?
时间: 2024-11-08 10:24:41 浏览: 35
针对偶氮苯分子及其衍生物在石墨烯表面的光储材料性能优化问题,推荐详细研究《优化光储材料:石墨烯-偶氮苯衍生物的分子模型与DFT计算》。该资料提供了关于如何通过第一性原理的密度泛函理论(DFT)计算来分析和设计偶氮苯分子模型,尤其是取代基的选择对材料性能的影响。
参考资源链接:[优化光储材料:石墨烯-偶氮苯衍生物的分子模型与DFT计算](https://wenku.csdn.net/doc/16ke2weeo8?spm=1055.2569.3001.10343)
DFT计算能够帮助我们预测分子的电子结构和能量状态,从而指导实验中取代基的选择,以优化偶氮苯分子的光致异构化性能和热稳定性。研究者可以通过构建分子模型,计算不同取代基的偶氮苯与石墨烯复合物的电子结构,评估顺反异构体之间的能量差异,以及分析光照下的反应动力学。
例如,通过对羧酸基、羟基、甲氧基、硝基等取代基的偶氮苯进行DFT计算,我们可以找出使偶氮苯分子在光照下具有更高能量存储能力的特定取代基。同时,通过计算评估不同取代基对材料热稳定性的贡献,可以筛选出能在高温环境下稳定保持结构的最优分子设计。
在掌握了DFT计算这一强大的工具后,研究者可以更精准地设计光储材料,不仅限于偶氮苯分子,还包括其他光化学反应中关键的分子。进一步的深入研究和实践应用,可参考《优化光储材料:石墨烯-偶氮苯衍生物的分子模型与DFT计算》来获取更多的理论与实验指导,推动光储材料领域的发展。
参考资源链接:[优化光储材料:石墨烯-偶氮苯衍生物的分子模型与DFT计算](https://wenku.csdn.net/doc/16ke2weeo8?spm=1055.2569.3001.10343)
阅读全文
相关推荐
大家在看
ccs中文教程
ccs的中文入门教程,很好的,讲的很详细
电路ESD防护原理与设计实例.pdf
电路ESD防护原理与设计实例,不错的资源,硬件设计参考,相互学习
计算机领域EI和SCI收录期刊、影响因子及国际会议
计算机领域EI和SCI收录期刊、影响因子及国际会议,文档中列出了计算机领域(无线通讯、微处理器、生物信息、数据无、数据挖掘和机器学习等)所有Rank1和Rank2级别的国际会议,网上给的资料一般都不全,好不容易找到,给大家分享一下,绝对值得下载!
HN8145XR-V5R021C00S260
HN8145XR_V5R021C00S260固件及V5使能工具等
赚分下文件
使用eclipse来写R程序
自己在安装StatET时,多次出错。所以写下这篇文档与大家分享。
最新推荐
DFT和FFT算法的比较
Good-Thomas算法,又称为Prime-Factor Algorithm (PFA),利用数论中的质因数分解将DFT转化为一系列较小的DFT和乘积,减少了乘法的复杂性。Rader质数因子算法是Good-Thomas算法的一种特殊情况,特别适合于N是质数的...
数字信号处理大作业1 利用DFT分析信号频谱
《数字信号处理大作业1:利用DFT分析信号频谱》 数字信号处理是一门涉及信息处理的关键技术,尤其在电子信息工程领域中占有重要地位。本篇大作业聚焦于使用离散傅里叶变换(DFT)来分析信号的频谱特性,并探讨不同...
数字信号处理实验报告-(2)-离散傅里叶变换(DFT).doc
实验中,使用MATLAB语言实现DFT和DFS的相关计算和图形化展示。给定一个序列主值x(n) = [0 1 2 3 2 1 0],实验目标包括: 1. 计算并显示该序列的幅度频谱和相位频谱,这可以通过执行DFT并提取复数结果的模和角度来...
DFT对称性的验证.doc
- 对于实数序列,利用DFT的对称性,可以通过构造复数序列并计算一次DFT,然后提取实部和虚部来高效计算两个实序列的DFT。这减少了计算量,尤其适用于FFT(快速傅立叶变换)算法。 - 当序列是纯虚数时,也有类似的...
HTML挑战:30天技术学习之旅
资源摘要信息: "desafio-30dias"
标题 "desafio-30dias" 暗示这可能是一个与挑战或训练相关的项目,这在编程和学习新技能的上下文中相当常见。标题中的数字“30”很可能表明这个挑战涉及为期30天的时间框架。此外,由于标题是西班牙语,我们可以推测这个项目可能起源于或至少是针对西班牙语使用者的社区。标题本身没有透露技术上的具体内容,但挑战通常涉及一系列任务,旨在提升个人的某项技能或知识水平。
描述 "desafio-30dias" 并没有提供进一步的信息,它重复了标题的内容。因此,我们不能从中获得关于项目具体细节的额外信息。描述通常用于详细说明项目的性质、目标和期望成果,但由于这里没有具体描述,我们只能依靠标题和相关标签进行推测。
标签 "HTML" 表明这个挑战很可能与HTML(超文本标记语言)有关。HTML是构成网页和网页应用基础的标记语言,用于创建和定义内容的结构、格式和语义。由于标签指定了HTML,我们可以合理假设这个30天挑战的目的是学习或提升HTML技能。它可能包含创建网页、实现网页设计、理解HTML5的新特性等方面的任务。
压缩包子文件的文件名称列表 "desafio-30dias-master" 指向了一个可能包含挑战相关材料的压缩文件。文件名中的“master”表明这可能是一个主文件或包含最终版本材料的文件夹。通常,在版本控制系统如Git中,“master”分支代表项目的主分支,用于存放项目的稳定版本。考虑到这个文件名称的格式,它可能是一个包含所有相关文件和资源的ZIP或RAR压缩文件。
结合这些信息,我们可以推测,这个30天挑战可能涉及了一系列的编程任务和练习,旨在通过实践项目来提高对HTML的理解和应用能力。这些任务可能包括设计和开发静态和动态网页,学习如何使用HTML5增强网页的功能和用户体验,以及如何将HTML与CSS(层叠样式表)和JavaScript等其他技术结合,制作出丰富的交互式网站。
综上所述,这个项目可能是一个为期30天的HTML学习计划,设计给希望提升前端开发能力的开发者,尤其是那些对HTML基础和最新标准感兴趣的人。挑战可能包含了理论学习和实践练习,鼓励参与者通过构建实际项目来学习和巩固知识点。通过这样的学习过程,参与者可以提高在现代网页开发环境中的竞争力,为创建更加复杂和引人入胜的网页打下坚实的基础。
【CodeBlocks精通指南】:一步到位安装wxWidgets库(新手必备)

# 摘要
本文旨在为使用CodeBlocks和wxWidgets库的开发者提供详细的安装、配置、实践操作指南和性能优化建议。文章首先介绍了CodeBlocks和wxWidgets库的基本概念和安装流程,然后深入探讨了CodeBlocks的高级功能定制和wxWidgets的架构特性。随后,通过实践操作章节,指导读者如何创建和运行一个wxWidgets项目,包括界面设计、事件
andorid studio 配置ERROR: Cause: unable to find valid certification path to requested target
### 解决 Android Studio SSL 证书验证问题
当遇到 `unable to find valid certification path` 错误时,这通常意味着 Java 运行环境无法识别服务器提供的 SSL 证书。解决方案涉及更新本地的信任库或调整项目中的网络请求设置。
#### 方法一:安装自定义 CA 证书到 JDK 中
对于企业内部使用的私有 CA 颁发的证书,可以将其导入至 JRE 的信任库中:
1. 获取 `.crt` 或者 `.cer` 文件形式的企业根证书;
2. 使用命令行工具 keytool 将其加入 cacerts 文件内:
```
VC++实现文件顺序读写操作的技巧与实践
资源摘要信息:"vc++文件的顺序读写操作"
在计算机编程中,文件的顺序读写操作是最基础的操作之一,尤其在使用C++语言进行开发时,了解和掌握文件的顺序读写操作是十分重要的。在Microsoft的Visual C++(简称VC++)开发环境中,可以通过标准库中的文件操作函数来实现顺序读写功能。
### 文件顺序读写基础
顺序读写指的是从文件的开始处逐个读取或写入数据,直到文件结束。这与随机读写不同,后者可以任意位置读取或写入数据。顺序读写操作通常用于处理日志文件、文本文件等不需要频繁随机访问的文件。
### VC++中的文件流类
在VC++中,顺序读写操作主要使用的是C++标准库中的fstream类,包括ifstream(用于从文件中读取数据)和ofstream(用于向文件写入数据)两个类。这两个类都是从fstream类继承而来,提供了基本的文件操作功能。
### 实现文件顺序读写操作的步骤
1. **包含必要的头文件**:要进行文件操作,首先需要包含fstream头文件。
```cpp
#include <fstream>
```
2. **创建文件流对象**:创建ifstream或ofstream对象,用于打开文件。
```cpp
ifstream inFile("example.txt"); // 用于读操作
ofstream outFile("example.txt"); // 用于写操作
```
3. **打开文件**:使用文件流对象的成员函数open()来打开文件。如果不需要在创建对象时指定文件路径,也可以在对象创建后调用open()。
```cpp
inFile.open("example.txt", std::ios::in); // 以读模式打开
outFile.open("example.txt", std::ios::out); // 以写模式打开
```
4. **读写数据**:使用文件流对象的成员函数进行数据的读取或写入。对于读操作,可以使用 >> 运算符、get()、read()等方法;对于写操作,可以使用 << 运算符、write()等方法。
```cpp
// 读取操作示例
char c;
while (inFile >> c) {
// 处理读取的数据c
}
// 写入操作示例
const char *text = "Hello, World!";
outFile << text;
```
5. **关闭文件**:操作完成后,应关闭文件,释放资源。
```cpp
inFile.close();
outFile.close();
```
### 文件顺序读写的注意事项
- 在进行文件读写之前,需要确保文件确实存在,且程序有足够的权限对文件进行读写操作。
- 使用文件流进行读写时,应注意文件流的错误状态。例如,在读取完文件后,应检查文件流是否到达文件末尾(failbit)。
- 在写入文件时,如果目标文件不存在,某些open()操作会自动创建文件。如果文件已存在,open()操作则会清空原文件内容,除非使用了追加模式(std::ios::app)。
- 对于大文件的读写,应考虑内存使用情况,避免一次性读取过多数据导致内存溢出。
- 在程序结束前,应该关闭所有打开的文件流。虽然文件流对象的析构函数会自动关闭文件,但显式调用close()是一个好习惯。
### 常用的文件操作函数
- `open()`:打开文件。
- `close()`:关闭文件。
- `read()`:从文件读取数据到缓冲区。
- `write()`:向文件写入数据。
- `tellg()` 和 `tellp()`:分别返回当前读取位置和写入位置。
- `seekg()` 和 `seekp()`:设置文件流的位置。
### 总结
在VC++中实现顺序读写操作,是进行文件处理和数据持久化的基础。通过使用C++的标准库中的fstream类,我们可以方便地进行文件读写操作。掌握文件顺序读写不仅可以帮助我们在实际开发中处理数据文件,还可以加深我们对C++语言和文件I/O操作的理解。需要注意的是,在进行文件操作时,合理管理和异常处理是非常重要的,这有助于确保程序的健壮性和数据的安全。
【大数据时代必备:Hadoop框架深度解析】:掌握核心组件,开启数据科学之旅
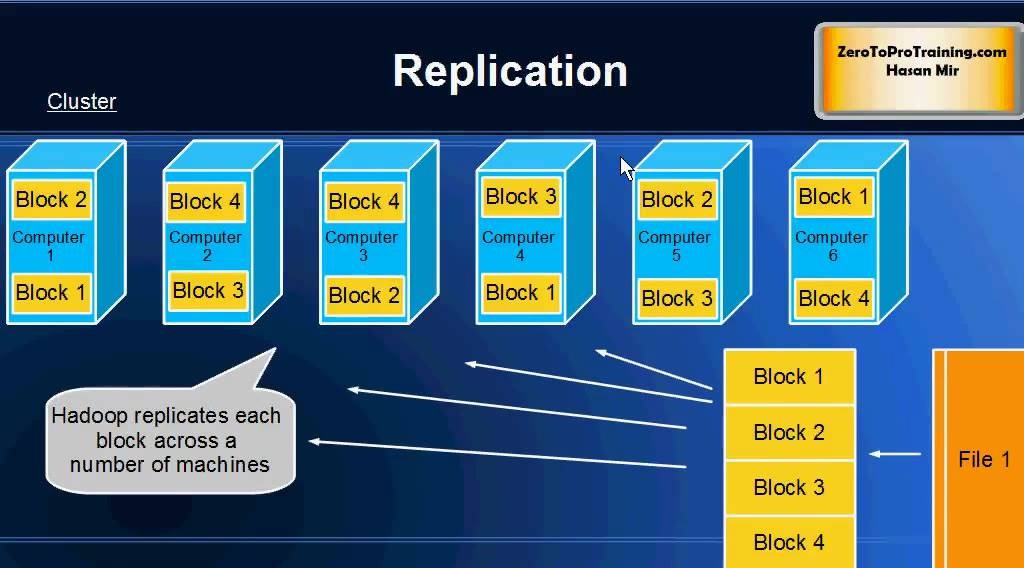
# 摘要
Hadoop作为一个开源的分布式存储和计算框架,在大数据处理领域发挥着举足轻重的作用。本文首先对Hadoop进行了概述,并介绍了其生态系统中的核心组件。深入分
opencv的demo程序
### OpenCV 示例程序
#### 图像读取与显示
下面展示如何使用 Python 接口来加载并显示一张图片:
```python
import cv2
# 加载图像
img = cv2.imread('path_to_image.jpg')
# 创建窗口用于显示图像
cv2.namedWindow('image', cv2.WINDOW_AUTOSIZE)
# 显示图像
cv2.imshow('image', img)
# 等待按键事件
cv2.waitKey(0)
# 销毁所有创建的窗口
cv2.destroyAllWindows()
```
这段代码展示了最基本的图