代码:r语言用for循环对一个基因表达数据框实现padj
时间: 2024-04-30 14:22:31 浏览: 73
以下是一个示例代码,用R语言的for循环对一个基因表达数据框实现padj:
```
# 假设我们有一个基因表达数据框,名为expr_df,其中包含基因名称、表达值和p值
# 表达值和p值均为数值型变量,基因名称为字符型
# 首先,我们需要将p值进行多重比较校正,这里使用Benjamini-Hochberg方法
# 这可以通过p.adjust函数实现
expr_df$padj <- p.adjust(expr_df$p, method = "BH")
# 接下来,我们将用for循环对每一行数据进行处理
for (i in 1:nrow(expr_df)) {
# 如果padj小于0.05,则将该基因标记为显著差异基因,否则标记为非显著差异基因
if (expr_df$padj[i] < 0.05) {
expr_df$diff[i] <- "yes"
} else {
expr_df$diff[i] <- "no"
}
}
# 最后,我们可以查看处理后的基因表达数据框
expr_df
```
在上述代码中,我们首先使用p.adjust函数将p值进行多重比较校正,然后使用for循环对每一行数据进行处理。在循环中,我们判断每个基因的padj值是否小于0.05,如果是则将该基因标记为显著差异基因,否则标记为非显著差异基因。最后,我们输出处理后的基因表达数据框。
相关问题
anotation<-function(node.list,genes,disease){ genes=genes[genes[,"disease"]%in%disease,] for (i in 1:nrow(node.list)){ if(node.list[i,"ENSG"]%in%genes[,"gene"]){ node.list[i,"padj"]=max(as.numeric(genes[genes[,"gene"]%in%node.list[i,"ENSG"],"padj"])) }else{ node.list[i,"padj"]=0 }} return(node.list) }
这段代码定义了一个名为 `anotation` 的函数,它接受三个参数 `node.list`、`genes` 和 `disease`。以下是对该函数的解释:
1. 首先,代码中的 `genes=genes[genes[,"disease"]%in%disease,]` 用于筛选出 `genes` 数据框中 "disease" 列的值与给定的 `disease` 向量中的值匹配的行。这将更新 `genes` 数据框,只保留与指定疾病相关的行。
2. 然后,代码使用 `for` 循环来遍历 `node.list` 数据框中的每一行。在循环中,使用条件语句 `if` 来判断 `node.list` 中的 "ENSG" 列的值是否存在于 `genes` 数据框中的 "gene" 列。
3. 如果 "ENSG" 值存在于 `genes` 数据框中的 "gene" 列,则执行以下代码块:
- 使用 `genes[genes[,"gene"] %in% node.list[i,"ENSG"],"padj"]` 从 `genes` 数据框中选择 "gene" 列值等于当前 "ENSG" 值的行,并提取这些行中 "padj" 列的最大值。
- 将最大值通过 `as.numeric()` 函数转换为数值类型,并赋值给 `node.list` 数据框中当前行的 "padj" 列。
4. 如果 "ENSG" 值不存在于 `genes` 数据框中的 "gene" 列,则将 `node.list` 数据框中当前行的 "padj" 列设置为 0。
5. 最后,函数返回更新后的 `node.list` 数据框。
这个函数似乎用于根据与指定疾病相关的基因信息,更新节点列表中的 "padj" 值。函数内部使用了条件判断、循环和数据框操作,但对于其他未显示的函数或数据结构,我们无法了解其具体实现细节。如果你需要更具体的帮助,请提供更多相关代码或详细说明。
阅读全文
相关推荐
大家在看
STM8L051F3P6使用手册(中文).zip
STM8L051
千方百剂服务器及客户端安装白皮书
千方百剂服务器及客户端安装白皮书.doc
ORACLE RMAN备份恢复指南
包含RMAN全量、增量、备份、恢复以及数据丢失、控制文件丢失、参数文件丢失、密码文件丢失、redo文件丢失、表空间损坏相关操作。
批量标准矢量shp互转txt工具
1.解压运行exe即可。(适用于windows7、windows10等操作系统)
2.标准矢量shp,转换为标准txt格式
4.此工具专门针对自然资源系统:建设用地报批、设施农用地上图、卫片等系统。
LTE软件使用介绍
基站管理工具的功能和特点 ,安装及配置使用方法。
最新推荐
ningyaozhongguogeshui
ningyaozhongguogeshui
时间控件,timer controller, 桌面小时间控件,简单的时间控件
时间控件,timer controller, 桌面小时间控件,简单的时间控件,
基于 DWT 的 STM32(或任何 ARM)的微秒级延迟库.zip
资源内项目源码是均来自个人的课程设计、毕业设计或者具体项目,代码都测试ok,都是运行成功后才上传资源,答辩评审绝对信服的,拿来就能用。放心下载使用!源码、说明、论文、数据集一站式服务,拿来就能用的绝对好资源!!!
项目备注
1、该资源内项目代码都经过测试运行成功,功能ok的情况下才上传的,请放心下载使用!
2、本项目适合计算机相关专业(如计科、人工智能、通信工程、自动化、电子信息等)的在校学生、老师或者企业员工下载学习,也适合小白学习进阶,当然也可作为毕设项目、课程设计、大作业、项目初期立项演示等。
3、如果基础还行,也可在此代码基础上进行修改,以实现其他功能,也可用于毕设、课设、作业等。 下载后请首先打开README.md文件(如有),仅供学习参考, 切勿用于商业用途。
4、如有侵权请私信博主,感谢支持
粒子群轨迹规划,3-5-3多项式时间最优轨迹规划,复现文章代码
粒子群轨迹规划,3-5-3多项式时间最优轨迹规划,复现文章代码
西门子1200PLC博途程序,博图版本V14及以上,具体为双行星动力搅拌桨混合机项目,有画面案例,硬件采用-S7-1200PLC加西门子KTP触摸屏 程序结构包括: 1.配料系统物料分
西门子1200PLC博途程序,博图版本V14及以上,具体为双行星动力搅拌桨混合机项目,有画面案例,硬件采用_S7-1200PLC加西门子KTP触摸屏。
程序结构包括:
1.配料系统物料分配-搅拌控制,分散控制
2.模拟量转,监测压力,称重,液位控制
3.PROFIBUS通讯监控电能表,搅拌电流监控
4.配方控制
5.变频器控制
6.高速计数器
硬件:
油泵电机:5.5KW
变频器:丹佛斯(丹麦)
PLC:西门子S7-1200
触摸屏:西门子KTP1200
海康无插件摄像头WEB开发包(20200616-20201102163221)
资源摘要信息:"海康无插件开发包"
知识点一:海康品牌简介
海康威视是全球知名的安防监控设备生产与服务提供商,总部位于中国杭州,其产品广泛应用于公共安全、智能交通、智能家居等多个领域。海康的产品以先进的技术、稳定可靠的性能和良好的用户体验著称,在全球监控设备市场占有重要地位。
知识点二:无插件技术
无插件技术指的是在用户访问网页时,无需额外安装或运行浏览器插件即可实现网页内的功能,如播放视频、音频、动画等。这种方式可以提升用户体验,减少安装插件的繁琐过程,同时由于避免了插件可能存在的安全漏洞,也提高了系统的安全性。无插件技术通常依赖HTML5、JavaScript、WebGL等现代网页技术实现。
知识点三:网络视频监控
网络视频监控是指通过IP网络将监控摄像机连接起来,实现实时远程监控的技术。与传统的模拟监控相比,网络视频监控具备传输距离远、布线简单、可远程监控和智能分析等特点。无插件网络视频监控开发包允许开发者在不依赖浏览器插件的情况下,集成视频监控功能到网页中,方便了用户查看和管理。
知识点四:摄像头技术
摄像头是将光学图像转换成电子信号的装置,广泛应用于图像采集、视频通讯、安全监控等领域。现代摄像头技术包括CCD和CMOS传感器技术,以及图像处理、编码压缩等技术。海康作为行业内的领军企业,其摄像头产品线覆盖了从高清到4K甚至更高分辨率的摄像机,同时在图像处理、智能分析等技术上不断创新。
知识点五:WEB开发包的应用
WEB开发包通常包含了实现特定功能所需的脚本、接口文档、API以及示例代码等资源。开发者可以利用这些资源快速地将特定功能集成到自己的网页应用中。对于“海康web无插件开发包.zip”,它可能包含了实现海康摄像头无插件网络视频监控功能的前端代码和API接口等,让开发者能够在不安装任何插件的情况下实现视频流的展示、控制和其他相关功能。
知识点六:技术兼容性与标准化
无插件技术的实现通常需要遵循一定的技术标准和协议,比如支持主流的Web标准和兼容多种浏览器。此外,无插件技术也需要考虑到不同操作系统和浏览器间的兼容性问题,以确保功能的正常使用和用户体验的一致性。
知识点七:安全性能
无插件技术相较于传统插件技术在安全性上具有明显优势。由于减少了外部插件的使用,因此降低了潜在的攻击面和漏洞风险。在涉及监控等安全敏感的领域中,这种技术尤其受到青睐。
知识点八:开发包的更新与维护
从文件名“WEB无插件开发包_20200616_20201102163221”可以推断,该开发包具有版本信息和时间戳,表明它是一个经过时间更新和维护的工具包。在使用此类工具包时,开发者需要关注官方发布的版本更新信息和补丁,及时升级以获得最新的功能和安全修正。
综上所述,海康提供的无插件开发包是针对其摄像头产品的网络视频监控解决方案,这一方案通过现代的无插件网络技术,为开发者提供了方便、安全且标准化的集成方式,以实现便捷的网络视频监控功能。
PCNM空间分析新手必读:R语言实现从入门到精通

# 摘要
本文旨在介绍PCNM空间分析方法及其在R语言中的实践应用。首先,文章通过介绍PCNM的理论基础和分析步骤,提供了对空间自相关性和PCNM数学原理的深入理解。随后,详细阐述了R语言在空间数据分析中的基础知识和准备工作,以及如何在R语言环境下进行PCNM分析和结果解
生成一个自动打怪的脚本
创建一个自动打怪的游戏脚本通常是针对游戏客户端或特定类型的自动化工具如Roblox Studio、Unity等的定制操作。这类脚本通常是利用游戏内部的逻辑漏洞或API来控制角色的动作,模拟玩家的行为,如移动、攻击怪物。然而,这种行为需要对游戏机制有深入理解,而且很多游戏会有反作弊机制,自动打怪可能会被视为作弊而被封禁。
以下是一个非常基础的Python脚本例子,假设我们是在使用类似PyAutoGUI库模拟键盘输入来控制游戏角色:
```python
import pyautogui
# 角色位置和怪物位置
player_pos = (0, 0) # 这里是你的角色当前位置
monster
CarMarker-Animation: 地图标记动画及转向库
资源摘要信息:"CarMarker-Animation是一个开源库,旨在帮助开发者在谷歌地图上实现平滑的标记动画效果。通过该库,开发者可以实现标记沿路线移动,并在移动过程中根据道路曲线实现平滑转弯。这不仅提升了用户体验,也增强了地图应用的交互性。
在详细的技术实现上,CarMarker-Animation库可能会涉及到以下几个方面的知识点:
1. 地图API集成:该库可能基于谷歌地图的API进行开发,因此开发者需要有谷歌地图API的使用经验,并了解如何在项目中集成谷歌地图。
2. 动画效果实现:为了实现平滑的动画效果,开发者需要掌握CSS动画或者JavaScript动画的实现方法,包括关键帧动画、过渡动画等。
3. 地图路径计算:标记在地图上的移动需要基于实际的道路网络,因此开发者可能需要使用路径规划算法,如Dijkstra算法或者A*搜索算法,来计算出最合适的路线。
4. 路径平滑处理:仅仅计算出路线是不够的,还需要对路径进行平滑处理,以使标记在转弯时更加自然。这可能涉及到曲线拟合算法,如贝塞尔曲线拟合。
5. 地图交互设计:为了与用户的交互更为友好,开发者需要了解用户界面和用户体验设计原则,并将这些原则应用到动画效果的开发中。
6. 性能优化:在实现复杂的动画效果时,需要考虑程序的性能。开发者需要知道如何优化动画性能,减少卡顿,确保流畅的用户体验。
7. 开源协议遵守:由于CarMarker-Animation是一个开源库,开发者在使用该库时,需要遵守其开源协议,合理使用代码并遵守贡献指南。
此库的文件名'CarMarker-Animation-master'表明这是一个主分支的项目,可能包含源代码文件、示例项目、文档说明等资源。开发者可以通过下载解压缩后获得这些资源,并根据提供的文档来了解如何安装和使用该库。在使用过程中,建议仔细阅读开源项目的贡献指南和使用说明,以确保库的正确集成和使用,同时也可以参与开源社区,与其他开发者共同维护和改进这一项目。"
5G核心网元性能瓶颈揭秘
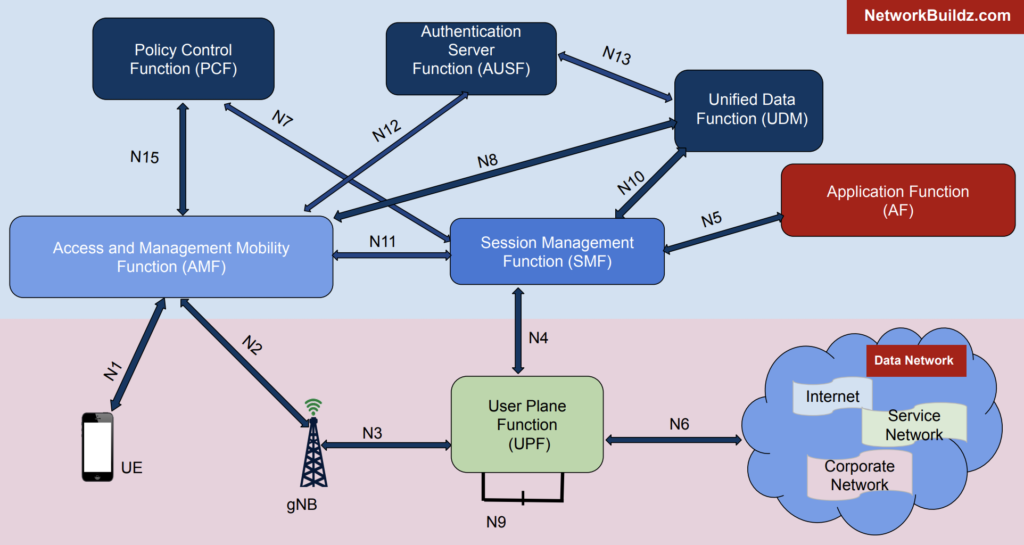
# 摘要
随着5G技术的发展和应用,其核心网的性能优化成为了行业关注的焦点。本文首先概述了5G核心网的架构,并对性能瓶颈进行深入分析,识别了关键的性能指标和瓶颈识别方法。通过案例分析,展示了核心网元常见的性能问题及其诊断和解决过程。随后,文章提出了多项性能优化策略,包括网络设计、系统配置调整以及新技术的应用。此外,本文探讨了安全挑战如何影响核心网的性能,