R语言在生物信息学中的应用与分析
发布时间: 2024-01-14 09:32:49 阅读量: 268 订阅数: 36 

R语言与BIOCONDUCTOR生物信息学应用_
# 1. 生物信息学简介
## 1.1 生物信息学的定义和基本概念
生物信息学是一门利用计算机科学和统计学方法研究生物学问题的学科。它通过收集、整理和分析生物学数据,帮助科学家理解生物学系统的复杂性。
生物信息学的基本概念包括:
- 序列分析:对DNA、RNA和蛋白质序列进行比对、注释和预测,以揭示其结构和功能。
- 基因组学:研究整个基因组的组成、结构、功能和演化,以及基因组中的遗传变异。
- 转录组学:通过测定RNA表达量来研究基因的转录水平和调控网络。
- 蛋白质组学:研究蛋白质的组成、结构、功能和相互作用,以及蛋白质组中的变异和修饰。
## 1.2 生物信息学在科学研究中的重要性
随着高通量测序技术的快速发展,生物学实验产生了海量的数据。生物信息学通过开发和应用算法和工具,可以更好地处理和分析这些数据,从而帮助科学家回答生物学上的复杂问题。
生物信息学在以下方面对科学研究起到了重要作用:
- 基因组学研究:通过比对和注释基因组数据,揭示基因功能和演化,帮助理解生物多样性和进化。
- 转录组学研究:通过对转录组数据的分析,探究基因调控网络,研究疾病机制。
- 蛋白质组学研究:通过研究蛋白质的组成和结构,揭示其功能和相互作用,为药物设计和治疗提供基础。
## 1.3 R语言在生物信息学中的地位和作用
R语言是一种广泛应用于数据分析和统计建模的开源编程语言。在生物信息学中,R语言因其丰富的数据处理和统计分析功能而备受青睐。
R语言在生物信息学中的地位和作用主要体现在以下几个方面:
- 数据可视化:R语言提供了强大的绘图和可视化功能,可以帮助科学家将生物数据转化为直观的图形,更好地理解数据和结果。
- 统计分析:R语言拥有丰富的统计分析函数和包,可以帮助科学家对生物数据进行探索性分析、假设检验和建模等统计分析操作。
- 数据处理:R语言具备灵活高效的数据处理能力,可以对大规模的生物数据进行导入、清洗、整理和转换,为后续的分析和挖掘提供基础。
- 机器学习:R语言中的机器学习算法和包能够帮助科学家构建生物信息学模型,从而实现基于数据的预测和分类。
R语言作为生物信息学的一种重要工具,为科学家提供了强大的数据分析能力,助力生物信息学研究的发展。
# 2. R语言基础知识
### 2.1 R语言的安装和配置
R语言是一种用于统计分析和数据可视化的开源编程语言。在进行生物信息学研究过程中,首先需要安装和配置R语言的开发环境。
#### 2.1.1 安装R语言
R语言的官方网站为[https://www.r-project.org/](https://www.r-project.org/),在该网站上可以下载适用于不同操作系统的R语言安装包。
对于Windows用户,可以按照以下步骤安装R语言:
1. 打开R语言官方网站,点击"CRAN"菜单项,选择适用于Windows的安装包下载链接。
2. 下载安装包后,双击运行安装包进行安装。
3. 在安装过程中,可以选择自定义安装路径和组件,根据个人需求进行选择并完成安装。
对于Mac用户,可以按照以下步骤安装R语言:
1. 打开R语言官方网站,点击"CRAN"菜单项,选择适用于Mac的安装包下载链接。
2. 下载安装包后,双击运行安装包进行安装。
3. 在安装过程中,按照提示进行操作并完成安装。
#### 2.1.2 配置R语言环境
安装完R语言之后,还需要进行一些配置,以便更好地使用R语言进行生物信息学分析。
1. 设置R语言的默认工作路径:
在R语言中,可以通过设置工作路径来指定R脚本和数据文件的存储位置。可以使用以下代码将工作路径设置为指定目录:
```R
setwd("/path/to/your/directory/")
```
2. 安装常用的生物信息学包:
R语言提供了许多用于生物信息学分析的扩展包。可以使用以下代码安装和加载常用的生物信息学包:
```R
install.packages("BiocManager")
BiocManager::install("Bioconductor")
```
3. 配置RStudio(可选):
RStudio是一款集成开发环境(IDE),提供了更便捷的R语言编程环境。可以根据个人需要下载并安装RStudio,然后在RStudio中编写和运行R代码。
### 2.2 R语言的基本语法和数据结构
#### 2.2.1 变量
在R语言中,可以使用`<-`或`=`符号来给变量赋值。例如:
```R
x <- 5
y = 10
```
#### 2.2.2 向量
R语言中的向量是存储相同类型元素的容器。可以使用`c()`函数创建向量。例如:
```R
numbers <- c(1, 2, 3, 4, 5)
```
#### 2.2.3 数据框
数据框是R语言中常用的数据结构,类似于Excel中的表格。可以使用`data.frame()`函数创建数据框。例如:
```R
df <- data.frame(name = c("Alice", "Bob", "Charlie"),
age = c(25, 30, 35),
gender = c("Female", "Male", "Male"))
```
### 2.3 R语言的常用函数和操作符
#### 2.3.1 常用函数
- `mean()`:计算向量或数据框中元素的平均值。
- `sum()`:计算向量或数据框中元素的和。
- `length()`:返回向量或数据框中元素的个数。
- `str()`:查看对象的结构和属性信息。
- `subset()`:根据条件筛选数据框的子集。
#### 2.3.2 常用操作符
- 算术操作符:`+`、`-`、`*`、`/`。
- 比较操作符:`<`、`>`、`==`、`!=`。
- 逻辑操作符:`&`(与)、`|`(或)、`!`(非)。
以上仅是R语言基础知识的简要介绍,接下来的章节将更加深入地介绍R语言在生物信息学中的各种应用和分析方法。
# 3. 生物数据的处理与分析
### 3.1 基因组数据的导入和清洗
在生物信息学研究中,处理和分析基因组数据是非常重要的一步。这些数据通常以文本文件的形式存在,包含了大量的基因组序列信息。而对这些数据进行正确的导入和清洗,可以为后续的分析提供准确的数据基础。
#### 3.1.1 导入基因组数据
R语言提供了丰富的包和函数,可以轻松导入各种格式的基因组数据。下面是一些常见的导入函数的使用示例:
```R
# 导入fasta格式的基因组数据
library(Biostrings)
genome_fasta <- readDNAStringSet("genome.fasta")
# 导入其他格式的基因组数据
library(GenomicFeatures)
genome_gff <- readGFF("genome.gff")
# 导入sam格式的比对数据
library(Rsamtools)
alignment_sam <- readBam("alignment.bam")
```
#### 3.1.2 清洗基因组数据
导入基因组数据后,需要进行数据清洗,以去除可能存在的错误或低质量的数据。常见的数据清洗操作包括:
- 去除低质量的序列:通过设置阈值并对质量进行筛选,去除质量较低的基因组序列。
- 去除冗余序列:通过比对和去除冗余序列,减少基因组数据的大小。
- 校正错误序列:通过比对和参考序列进行校正,纠正基因组数据中的错误。
下面是一个清洗基因组数据的示例:
```R
# 去除低质量的序列
filtered_genome <- genome_fasta[quality(genome_fasta) > 20]
# 去除冗余序列
deduplicated_genome <- unique(filtered_genome)
# 校正错误序列
corrected_genome <- alignAndCorrect(deduplicated_genome, reference_genome)
```
### 3.2 序列比对与测序数据处理
序列比对是生物信息学中常见的任务之一,它可以将测序数据与参考基因组进行比对,从而确定测序reads的位置和对应的基因信息。R语言提供了多种包和函数,用于进行序列比对和测序数据的处理。
#### 3.2.1 序列比对
```R
# 导入比对算法的包
library(Bioconductor)
library(GenomicAlignments)
# 执行序列比对
aligned_reads <- alignSeq(sequences, reference_genome)
```
#### 3.2.2 测序数据处理
```R
# 导入测序数据处理的包
library(ShortRead)
library(RNAseqData.HNRNPC.bam.chr14)
# 执行测序数据处理
cleaned_reads <- cleanFastq(reads)
```
### 3.3 基因表达谱分析
基因表达谱分析是生物信息学中常见的任务之一,它可以帮助研究人员了解不同基因在不同组织或条件下的表达水平。R语言提供了多种包和函数,用于进行基因表达谱分析。
```R
# 导入表达数据
expression_data <- read.table("expression_data.txt")
# 统计表达数据
mean_expression <- apply(expression_data, 1, mean)
# 绘制表达谱图
barplot(mean_expression, names.arg = rownames(expression_data), xlab = "Genes", ylab = "Expression")
```
### 3.4 DNA/RNA序列分析
R语言也可以用于DNA/RNA序列的分析,比如序列特征分析、序列比对和突变检测等。
```R
# 导入序列分析包
library(Biostrings)
# 分析序列特征
sequence <- DNAString("ATCGATCGA")
gc_content <- letterFrequency(sequence, letters = c("A", "T", "C", "G"))
# 序列比对和突变检测
reference_sequence <- DNAString("ATCGATCGA")
query_sequence <- DNAString("ATAGATCGA")
alignment <- pairwiseAlignment(reference_sequence, query_sequence)
mismatch_count <- countMismatches(alignment)
```
以上仅是基因组数据处理与分析的一部分示例,R语言在生物信息学中的应用非常广泛。根据具体的研究问题和数据类型,还可以使用其他的R包和函数进行更复杂的分析。
# 4. 生物图形技术与可视化
生物信息学中的数据可视化对于展现生物学数据和分析结果至关重要。R语言提供了丰富的绘图函数和包,可以用于绘制各种生物学图形和可视化结果。
### 4.1 绘制常见生物学图形的方法和技巧
在生物信息学中,常见的图形包括基因表达谱热图、柱状图、折线图、散点图、箱线图等。使用R语言的ggplot2、heatmap和其他绘图包可以轻松实现这些图形的绘制,通过调整参数和添加标签、标题等可以美化图形并准确传达数据信息。
```R
# 使用ggplot2绘制基因表达谱热图
library(ggplot2)
# 生成模拟数据
gene_data <- data.frame(
gene = c("Gene1", "Gene2", "Gene3", "Gene4"),
sample1 = c(3.2, 4.5, 2.7, 5.1),
sample2 = c(2.8, 3.9, 2.5, 4.9),
sample3 = c(3.0, 4.1, 2.6, 5.0)
)
# 转换数据格式为长格式
gene_data_long <- tidyr::gather(gene_data, "sample", "expression", -gene)
# 绘制热图
ggplot(gene_data_long, aes(x = sample, y = gene, fill = expression)) +
geom_tile(color = "white") +
scale_fill_gradient(low = "white", high = "steelblue") +
theme_minimal() +
labs(title = "Gene Expression Heatmap", x = "Sample", y = "Gene")
```
### 4.2 数据可视化在生物信息学中的应用
除了基因表达谱热图外,生物信息学中还经常使用箱线图展示基因表达差异、散点图展示样本间关系、柱状图展示基因注释信息等。这些图形的优雅绘制和清晰传达是生物信息学研究中不可或缺的一部分。
### 4.3 制作生物通路图和基因网络图
生物通路图和基因网络图是生物信息学研究中常用的图形,用于展示基因间相互作用和信号通路。R语言中的pathview和igraph包提供了绘制生物通路图和基因网络图的功能,通过这些工具可以清晰展示生物学数据和分析结果的复杂关系。
以上是关于生物图形技术与可视化的部分内容,通过对生物学数据的有效可视化,研究人员可以更好地理解数据、发现规律并进行更深入的分析。
# 5. 生物统计学与机器学习分析
#### 5.1 生物统计学在RNA-seq分析中的应用
生物统计学是生物信息学中非常重要的一部分,它主要用于对生物数据进行分析和解释。在RNA-seq分析中,生物统计学的应用尤为突出。下面将介绍一些常见的生物统计学方法和其在RNA-seq分析中的应用。
##### 5.1.1 差异表达基因分析
差异表达基因分析是RNA-seq中的关键步骤之一,通过比较不同条件下的基因表达水平,找出差异表达的基因。常见的差异表达分析方法包括DESeq2、edgeR和limma等。以DESeq2为例,下面是一个使用DESeq2进行差异表达基因分析的示例代码:
```R
# 导入DESeq2包
library(DESeq2)
# 读入原始表达数据
countData <- read.table("counts_data.txt", header = TRUE, row.names = 1)
# 创建DESeq2对象
dds <- DESeqDataSetFromMatrix(countData = countData, colData = colData, design = ~ condition)
# 进行差异表达分析
dds <- DESeq(dds)
# 获取差异表达基因
res <- results(dds)
# 对结果进行筛选和可视化
resSig <- subset(res, padj < 0.05 & abs(log2FoldChange) > 1)
plotMA(res, ylim = c(-5, 5))
```
##### 5.1.2 聚类分析
聚类分析是将相似的样本或基因分组在一起的分析方法,可帮助我们发现样本间或基因间的相似性和差异性。常用的聚类分析方法有层次聚类和k均值聚类等。下面是一个使用hclust进行层次聚类分析的示例代码:
```R
# 读入表达矩阵
exprMatrix <- read.table("expression_matrix.txt", header = TRUE, row.names = 1)
# 计算样本间的距离
distMatrix <- dist(t(exprMatrix))
# 执行层次聚类
hc <- hclust(distMatrix)
# 绘制聚类结果的热图
heatmap(exprMatrix, Colv = NA, Rowv = hc$order, scale = "row")
```
##### 5.1.3 Gene Ontology (GO) 富集分析
GO富集分析是一种用于确定不同基因集中的显著富集功能的统计分析方法。它可以帮助我们理解差异表达基因的功能和参与的生物学过程。常见的GO富集分析方法有GOstats和clusterProfiler等。以clusterProfiler为例,下面是一个使用clusterProfiler进行GO富集分析的示例代码:
```R
# 导入clusterProfiler包
library(clusterProfiler)
# 导入差异表达基因集
geneList <- read.table("differentially_expressed_genes.txt", header = TRUE)
# 进行GO富集分析
ego <- enrichGO(gene = geneList,
universe = allGenes,
keyType = "SYMBOL",
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
qvalueCutoff = 0.1,
readable = TRUE)
# 打印富集结果
print(ego)
```
#### 5.2 常用的机器学习算法及其在生物信息学中的应用
机器学习算法在生物信息学中的应用越来越广泛,可以用于预测蛋白质结构、基因寻找、序列标识和分类等任务。下面介绍几种常用的机器学习算法及其在生物信息学中的应用。
##### 5.2.1 支持向量机 (Support Vector Machine, SVM)
支持向量机是一种常用的机器学习算法,可以用于分类和回归任务。在生物信息学中,支持向量机可以应用于预测蛋白质的功能、预测基因家族和预测药物效果等任务。
```R
# 导入e1071包
library(e1071)
# 读入训练集和测试集
trainData <- read.table("train_data.txt", header = TRUE)
testData <- read.table("test_data.txt", header = TRUE)
# 创建SVM模型
svmModel <- svm(Class ~ ., data = trainData)
# 预测
svmPred <- predict(svmModel, testData)
```
##### 5.2.2 随机森林 (Random Forest)
随机森林是一种基于决策树的集成学习算法,可以用于分类和回归任务。在生物信息学中,随机森林可以应用于基因表达谱分类和特征选择等任务。
```R
# 导入randomForest包
library(randomForest)
# 读入训练集和测试集
trainData <- read.table("train_data.txt", header = TRUE)
testData <- read.table("test_data.txt", header = TRUE)
# 创建随机森林模型
rfModel <- randomForest(Class ~ ., data = trainData)
# 预测
rfPred <- predict(rfModel, testData)
```
##### 5.2.3 深度学习算法
深度学习算法在生物信息学中的应用也越来越广泛,可以用于图像识别、序列分类和蛋白质结构预测等任务。常见的深度学习算法有卷积神经网络 (Convolutional Neural Network, CNN)、循环神经网络 (Recurrent Neural Network, RNN) 和生成对抗网络 (Generative Adversarial Network, GAN) 等。
```R
# 导入keras包
library(keras)
# 创建CNN模型
model <- keras_model_sequential()
# 构建模型结构
# 编译模型
model %>% compile(
loss = 'categorical_crossentropy',
optimizer = optimizer_adadelta(),
metrics = c('accuracy')
)
# 训练模型
model %>% fit(
x_train, y_train,
epochs = 10,
batch_size = 128,
validation_split = 0.2
)
# 预测
pred <- model %>% predict_classes(x_test)
```
#### 5.3 基于R语言的生物信息学数据分析案例
在这一节,我们将通过一个基因表达谱分析的案例来展示R语言在生物信息学中的应用。案例的具体内容是针对一个白血病数据集,通过差异表达分析和GO富集分析来找出与白血病相关的基因和生物学过程。以下是案例示例代码的摘要:
```R
# 读入表达矩阵和样本信息
expressionMatrix <- read.table("expression_matrix.txt", header = TRUE, row.names = 1)
sampleInfo <- read.table("sample_info.txt", header = TRUE)
# 创建DESeq2对象
dds <- DESeqDataSetFromMatrix(countData = expressionMatrix, colData = sampleInfo, design = ~ condition)
# 进行差异表达分析
dds <- DESeq(dds)
# 获取差异表达基因
res <- results(dds)
differentiallyExpressedGenes <- subset(res, padj < 0.05 & abs(log2FoldChange) > 1)
# 进行GO富集分析
geneList <- rownames(differentiallyExpressedGenes)
ego <- enrichGO(gene = geneList,
universe = allGenes,
keyType = "SYMBOL",
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
qvalueCutoff = 0.1,
readable = TRUE)
# 打印富集结果
print(ego)
```
通过上述代码,我们可以对白血病数据进行差异表达分析,并找出与白血病相关的基因和生物学过程。这些结果可以帮助研究人员深入理解白血病的发生机制和寻找治疗靶点。
以上就是生物统计学与机器学习分析的章节内容,通过使用R语言中的生物信息学工具和算法,可以有效地处理和分析生物数据,从而取得研究的突破和发展。
# 6. R语言在生物信息学研究中的挑战与展望
生物信息学作为一个跨学科的领域,不断涌现出新的技术和方法。R语言作为一种强大的统计分析工具,在生物信息学研究中扮演着重要的角色。然而,R语言在生物信息学研究中也面临一些挑战。
### 6.1 R语言在生物信息学中的优点和局限性
#### 6.1.1 优点
- **丰富的生物信息学函数库:** R语言拥有众多的生物信息学函数库,可用于处理生物数据、进行统计分析和绘制图形。这些函数库能够满足生物信息学研究的需求。
- **开源和免费:** R语言是开源的,可以免费获取和使用。这使得研究人员可以更容易地使用R语言进行生物信息学研究,降低了研究成本。
- **灵活性和可扩展性:** R语言具有灵活性和可扩展性,可以通过编写自定义函数和使用第三方包来满足不同的研究需求。
#### 6.1.2 局限性
- **学习曲线陡峭:** 对于初学者来说,学习R语言可能需要一些时间和精力。由于其语法和数据结构与其他编程语言有所不同,初学者可能需要花费一些时间来适应和掌握。
- **运行效率相对较低:** 相比其他编程语言(如Python或C++),R语言在运行效率方面可能较低。这对处理大规模数据集或需要进行复杂计算的情况可能会产生一些挑战。
- **与其他分析平台的整合:** 在与其他分析平台或语言进行整合时,可能需要进行特定的数据格式转换和处理,这可能会增加一些额外的工作量和复杂性。
### 6.2 生物信息学研究中的新兴技术和趋势
生物信息学研究领域正在不断涌现出新的技术和趋势,这些将影响R语言在生物信息学中的应用。
- **单细胞测序技术:** 随着单细胞测序技术的发展,研究者可以对单个细胞进行基因表达谱分析。这为了解细胞类型、发育过程和疾病发展提供了更详细的信息。R语言具有丰富的单细胞测序分析工具和包,可以应对这一新兴技术的挑战。
- **深度学习在生物信息学中的应用:** 深度学习在图像分析和自然语言处理等领域取得了巨大的成功。在生物信息学中,深度学习可用于预测蛋白质结构、DNA/RNA序列分析等。R语言中已经有一些用于生物信息学的深度学习包,使研究者能够进行相关分析。
### 6.3 如何进一步提高R语言在生物信息学中的应用和效果
为了进一步提高R语言在生物信息学研究中的应用和效果,可以采取以下措施:
- **加强培训和教育:** 提供更多的培训和教育机会,帮助研究者掌握R语言的技能和知识,提高其在生物信息学中的应用水平。
- **改进R语言的运行效率:** 发展更高效的算法和方法,改进R语言在处理大规模数据和复杂计算方面的性能。
- **与其他编程语言和平台整合:** 提供更方便的工具和方法,帮助研究者将R语言与其他编程语言和平台进行整合,以满足不同分析需求。
- **丰富R语言的生物信息学函数库:** 开发和完善更多的生物信息学函数库和包,以支持更广泛的生物信息学研究。
通过以上措施的实施,可以进一步推动R语言在生物信息学中的应用和发展,为生物信息学研究提供更多有力的工具和方法。

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
本专栏将重点介绍生物数据分析与信息处理技术,涵盖了生物数据分析的基础概念与应用、Python和R语言在生物数据处理中的基本操作与实践、生物数据中的统计学基础与应用技巧、生物信息学中的序列比对与序列分析方法、生物图像处理技术的原理与实践、生物信息学中的机器学习算法及生物数据应用等多个方面。此外,我们还将讨论生命科学中的网络分析与生物大数据挖掘、基因组学数据分析的关键技术与方法探讨、生物数据清洗与预处理的常用技巧与工具、药物开发中的生物信息学方法与应用案例、蛋白质组学数据分析的基本原理与实践等内容。同时,我们还将深入探讨转录组数据分析的常用工具与技术,基因组序列数据挖掘与注释方法,生物信息学中的差异表达分析技术与实例讲解,以及环境基因组学数据分析的挑战与解决方案等领域。最后,我们将介绍基于深度学习的生物数据分析与应用,以及生物信息学中的高通量数据处理技术与案例研究。无论是对于生物信息学初学者还是专业人士来说,这个专栏都将提供丰富的信息和实用的技术,帮助读者更好地理解和应用生物数据分析与信息处理技术。
专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
深入解析WinPcap:网络数据包捕获机制与优化技巧
# 摘要
WinPcap作为一个广泛使用的网络数据包捕获库,为网络应用开发提供了强大的工具集。本文首先介绍了WinPcap的基本概念和安装配置方法,然后深入探讨了网络数据包捕获的基础知识,包括数据链路层与网络层解析,以及过滤器的原理与应用。接着,文章针对高级数据处理,阐述了数据包动态捕获、分析、统计和协议分析的方法,并提供了错误处理与调试的技巧。在实践章节
【MySQL性能优化】:从新手到专家的10大调整指南

# 摘要
本文详细探讨了MySQL数据库性能优化的各个方面,从基础架构到高级技术应用。首先介绍MySQL的性能优化理论基础,涵盖存储引擎、查询缓存、连接管理等关键组件,以及索引和SQL查询的优化策略。接着,文章转向性能监控和分析,讨论了性能监控工具、性能
【通信原理与2ASK系统的融合】:理论应用与实践案例分析

# 摘要
本论文首先对通信原理进行了概述,并详细探讨了2ASK(Amplitude Shift Keying)系统的理论基础,包括2ASK调制技术原理、性能分析、带宽需求以及硬件和软件实现。接着,通过多个应用场景,如无线通信、光通信和数字广播系统,分析了2ASK技术的实际应用和案例。文章还展望了通信系统技术的最新进展,探讨了2ASK技术的改进、创新及与其他技
【DeltaV OPC服务器深度优化】:数据流与同步的极致操控

# 摘要
本文系统性地介绍了DeltaV OPC服务器的基础知识、性能调优、高级功能实现以及未来发展趋势。首先,概述了DeltaV OPC服务器的基本概念和数据流同步机制。其次,深入探讨了性能调优的实践,包括系统配置和网络环境的影响,以及基于案例的性能提升分析。此外,本文还阐述了DeltaV OPC服务器在多
Jpivot大数据攻略:处理海量数据的12个策略
# 摘要
随着大数据时代的到来,Jpivot大数据处理的效率与质量成为企业和研究机构关注的焦点。本文概述了大数据处理的整体流程,从数据采集与预处理的策略制定,到海量数据的存储与管理,再到利用分布式计算框架进行数据分析与挖掘,最后通过数据可视化与报告展现结果并注重数据安全与隐私保护。通过对Jpivot大数据处理各阶段关键技术的
Altium Designer新手必读:函数使用全攻略
# 摘要
Altium Designer是一款广泛使用的电子设计自动化软件,其强大的函数功能是提高设计效率和实现设计自动化的关键。本文旨在对Altium Designer中的函数概念、类型、应用以及高级技巧进行系统性介绍。首先,概述了Altium Designer的基本函数基础,包括函数的定义、作用、常见类型以及内置和自定义函数的使用。随后,深入探讨了高级函数应用技巧,如参数传递、变量作用域、
Qt事件处理机制深入剖析

# 摘要
Qt框架以其跨平台特性和强大的事件处理机制,被广泛应用于GUI开发。本文深入探讨了Qt中的事件处理概念、理论基础以及实践技巧。从事件驱动编程模型到事件机制的理论基础,再到具体的编程实践,本文详细解析了Qt事件处理的各个方面。同时,文章深入分析了信号槽机制与事件之间的协同工作,并探讨了在Qt中实现异步事件处理、性能优化和跨平台兼容性的高级应用。通过对不同场景下的事件处理案例进行分析,本文总结了Qt事
PNOZ继电器应用优化:提高系统安全性能的实用技巧
# 摘要
PNOZ继电器是一种广泛应用于工业安全领域的关键设备,它通过一系列安全功能和特性来确保系统安全。本文详细介绍了PNOZ继电器的应用原理、在系统安全中的作用,以及与其他安全设备的协同工作。文章还探讨了继电器的配置与调试,优化实践,以及在不同行业中应用案例,以实现提升系统响应速度、稳定性和可靠性的目标。最后,本文展望了PNOZ继电器的未来发展趋势,关注新技术的融合和行业规范更新对继电器应
PN532 NFC芯片深度解析:从基础到应用
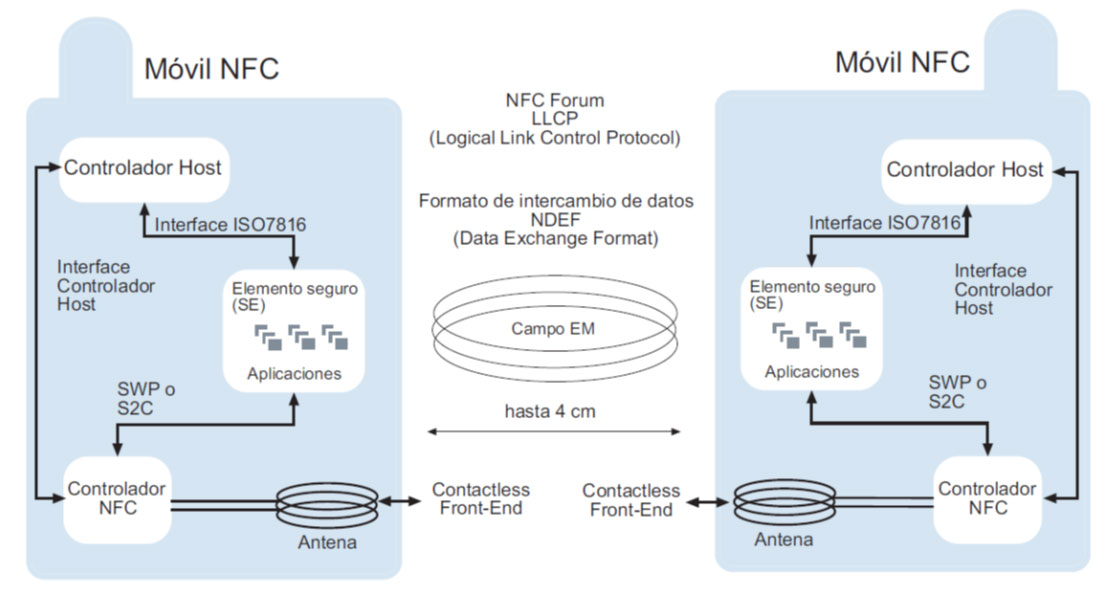
# 摘要
PN532 NFC芯片作为一款广泛应用于短距离无线通信的芯片,支持多种硬件接口和NFC通信协议。本文首先介绍了PN532 NFC芯片的基础特性,然后详细解析了其硬件接口如I2C、SPI、UART和HSU,以及NFC技术标准和通信模式。接着,文章转向编程基础,包括固件安装、配置寄存器和命令集,以及对不同类型NFC卡的读写操作实例。此外,文中还探
【故障诊断与预防】:LAT1173同步失败原因分析及预防策略

# 摘要
本文针对LAT1173同步失败现象进行了全面概述,深入探讨了其同步机制和理论基础,包括工作原理、同步过程中的关键参数以及同步失败模式和成功率影响因素。通过具体案例研究,本文剖析了硬件与软件层面导致同步失败的原因,并提出了一系列针对性的预防策略和故障处理措施。研究重点在于硬件维护升级和软件配置管理的最佳实践,旨在减少同步失败的风险,确保系统的稳定性和可靠性。
# 关键字
同步失败;理论分析;案例研究;故障预防;硬件维护;软件管理
参考资源链接:[STM3
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )