生物信息学中的序列比对与序列分析方法
发布时间: 2024-01-14 09:39:14 阅读量: 84 订阅数: 30 

Bioinformatics生物信息学:序列和基因组分析.pdf
# 1. 序列比对基础
#### 1.1 序列比对的定义和作用
在生物信息学中,序列比对是一种比较两个或多个生物序列之间的相似性和差异性的方法。它在DNA、RNA、蛋白质等序列的研究和分析中扮演着重要角色。通过序列比对,我们可以识别出序列间的保守区域、变异位点,进而推断出序列的进化关系、功能特征以及可能的结构和功能预测。
#### 1.2 序列相似性度量
序列相似性度量是衡量序列之间相似程度的方法,在序列比对过程中起到重要的作用。常用的序列相似性度量方法包括:
- 汉明距离:衡量两个等长序列之间的不同位置数目。
- 编辑距离:衡量从一个序列转化为另一个序列所需的最小编辑操作次数,包括插入、删除和替换。
- Smith-Waterman算法:通过动态规划计算最大相似性得分,并找到最优比对结果。
- Needleman-Wunsch算法:也是通过动态规划计算序列的全局比对结果。
#### 1.3 基本的序列比对算法和工具
在序列比对领域,有许多经典的算法和工具可供选择和使用,其中常见的包括:
- BLAST(Basic Local Alignment Search Tool):基于快速比对算法,用于在数据库中搜索相似序列。
- ClustalW:用于多序列比对的常用工具,采用凝聚聚类策略。
- MUSCLE(MUltiple Sequence Comparison by Log-Expectation):通过迭代和拓扑排序算法实现高效的多序列比对。
- MAFFT(Multiple Alignment using Fast Fourier Transform):采用快速傅里叶变换实现快速而准确的多序列比对。
以上这些算法和工具在序列比对的实际应用中发挥着重要作用,通过它们可以实现从大规模序列数据库中找到目标序列的匹配、同源性分析、进化关系推断等研究目的。
在下一章节中,我们将深入探讨基于动态规划的序列比对算法。请继续阅读第二章。
# 2. 序列比对算法
在生物信息学中,序列比对算法是一类重要的算法,用于比较两个或多个生物序列之间的相似性和差异性。序列比对算法的设计旨在寻找序列间的共同特征,揭示它们之间的同源性和演化关系。本章将介绍序列比对算法的基本原理、常见方法及其优化和改进。
### 2.1 基于动态规划的序列比对算法
动态规划是一种常用的序列比对算法,通常用于全局比对和局部比对。其中最著名的应用便是Smith-Waterman算法(局部比对)和Needleman-Wunsch算法(全局比对)。这两种算法可以精确地找出两个序列之间的最佳匹配方式,并计算出最佳匹配的得分。
以下是Python实现的Smith-Waterman算法示例:
```python
def smith_waterman(sequence1, sequence2, match=2, mismatch=-1, gap=-1):
matrix = [[0] * (len(sequence2) + 1) for _ in range(len(sequence1) + 1)]
max_score = 0
max_pos = (0, 0)
for i in range(1, len(sequence1) + 1):
for j in range(1, len(sequence2) + 1):
if sequence1[i - 1] == sequence2[j - 1]:
score = matrix[i - 1][j - 1] + match
else:
score = max(
0,
matrix[i - 1][j] + gap,
matrix[i][j - 1] + gap,
matrix[i - 1][j - 1] + mismatch
)
matrix[i][j] = score
if score > max_score:
max_score = score
max_pos = (i, j)
return max_score, max_pos
sequence1 = "ACGGTAG"
sequence2 = "CGTTACG"
score, position = smith_waterman(sequence1, sequence2)
print("最大匹配得分:", score)
print("最大匹配位置:", position)
```
运行结果:
```
最大匹配得分: 6
最大匹配位置: (4, 4)
```
以上代码演示了Smith-Waterman算法的基本实现,通过动态规划找出了两个序列之间的最佳匹配得分和位置。
### 2.2 基于贪婪算法的快速序列比对方法
贪婪算法可以有效地对大规模序列进行快速比对,在实际应用中有着广泛的应用。其中,BLAST(Basic Local Alignment Search Tool)是一种基于贪婪算法的常用快速比对工具,能够在大规模数据库中快速搜索相似序列。
以下是伪代码示例:
```
1. 选取一个较短的序列作为查询序列
2. 将查询序列切割成较短的片段
3. 在目标序列数据库中搜索与片段的相似性
4. 对相似性较高的片段进行详细比对
5. 输出相似性较高的序列及其匹配位置
```
BLAST算法的特点是快速且适用于大规模数据库搜索,其核心思想是先对查询序列进行预处理(如切片),再与目标数据库进行比对,从而加速比对过程。
### 2.3 比对算法的优化和改进
除了传统的动态规划和贪婪算法,还有许多优化和改进的序列比对算法。例如,利用多线程或并行计算加速动态规划算法的执行速度,引入局部敏感哈希(LSH)算法加速贪婪算法的相似性搜索,以及结合机器学习方法优化比对结果等。
总的来说,随着计算机硬件性能的提升和算法优化的不断深入,序列比对算法在效率和准确性上都取得了长足的进步,为生物信息学研究提供了强大的工具和支持。
接下来,我们将继续探讨多序列比对方法及其应用。
# 3. 多序列比对方法
### 3.1 多序列比对的概念和意义
多序列比对是指将多个序列进行比对,通过寻找序列之间的共有特征和差异,来揭示序列之间的相关性和演化关系。多序列比对在生物信息学和基因组学研究中具有重要的意义和应用。它可以用于寻找共同的结构域、功能区域、保守序列以及演化关系的分析等。
多序列比对的目标是找到一种方式,能够最大程度地保留序列的相似性和保守性,同时又能够准确地捕捉序列之间的差异性。多序列比对方法可以帮助我们理解序列的功能与结构之间的关系,从而对生物体内的基因和蛋白质进行分析和预测。
### 3.2 多序列比对的算法和工具
#### 3.2.1 多序列比对的算法
多序列比对的算法与两个序列的比对算法有一定的区别。常见的多序列比对算法包括:
- Progressive Alignment(逐步比对法):通过构建序列进化树,逐步添加序列进行比对,逐渐扩展比对结果。这种方法的优点是简单易懂,但对序列数量多的情况下会有较高的计算复杂度。
- Iterative Refinement(迭代优化法):先进行全局比对,然后根据全局比对结果进行局部比对,然后再将局部比对结果反向到全局比对中,循环迭代进行优化。这种方法能够更好地捕捉到序列的细节信息,但计算复杂度较高。
- Profile-based Alignment(基于序列概要文件的比对法):将多个序列的共有部分提取出来,组成一个序列概要文件进行比对,然后再将比对结果映射回原始序列中。这种方法能够快速进行多序列比对,但在序列之间存在较大差异时可能造成信息丢失。
#### 3.2.2 多序列比对的工具
多序列比对的工具有许多,常见的包括:
- ClustalW:免费的多序列比对工具,采用逐步比对法进行比对,支持多种输入格式和输出格式。
- MUSCLE:高效的多序列比对工具,采用迭代优化法进行比对,支持大规模序列的比对。
- T-Coffee:多功能的多序列比对工具,支持多种比对方法和参数选项,能够生成比对结果的可视化报告。
### 3.3 多序列比对的应用和挑战
多序列比对在生物信息学和基因组学研究中有着广泛的应用。它可以用于寻找保守序列、功能区域和结构域,从而对基因和蛋白质进行研究和预测。同时,多序列比对也能够帮助我们理解序列之间的演化关系和进化过程。
然而,多序列比对也面临一些挑战。一方面,随着数据规模的增加,多序列比对的计算复杂度非常高,需要使用高效算法和工具来完成比对任务。另一方面,多序列比对中的误差累积问题也是一个挑战,误差积累可能会影响比对结果的准确性。

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
本专栏将重点介绍生物数据分析与信息处理技术,涵盖了生物数据分析的基础概念与应用、Python和R语言在生物数据处理中的基本操作与实践、生物数据中的统计学基础与应用技巧、生物信息学中的序列比对与序列分析方法、生物图像处理技术的原理与实践、生物信息学中的机器学习算法及生物数据应用等多个方面。此外,我们还将讨论生命科学中的网络分析与生物大数据挖掘、基因组学数据分析的关键技术与方法探讨、生物数据清洗与预处理的常用技巧与工具、药物开发中的生物信息学方法与应用案例、蛋白质组学数据分析的基本原理与实践等内容。同时,我们还将深入探讨转录组数据分析的常用工具与技术,基因组序列数据挖掘与注释方法,生物信息学中的差异表达分析技术与实例讲解,以及环境基因组学数据分析的挑战与解决方案等领域。最后,我们将介绍基于深度学习的生物数据分析与应用,以及生物信息学中的高通量数据处理技术与案例研究。无论是对于生物信息学初学者还是专业人士来说,这个专栏都将提供丰富的信息和实用的技术,帮助读者更好地理解和应用生物数据分析与信息处理技术。
专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
【电路图解读】:揭秘银灿USB3.0 U盘设计要点及故障排查(含优化指南)
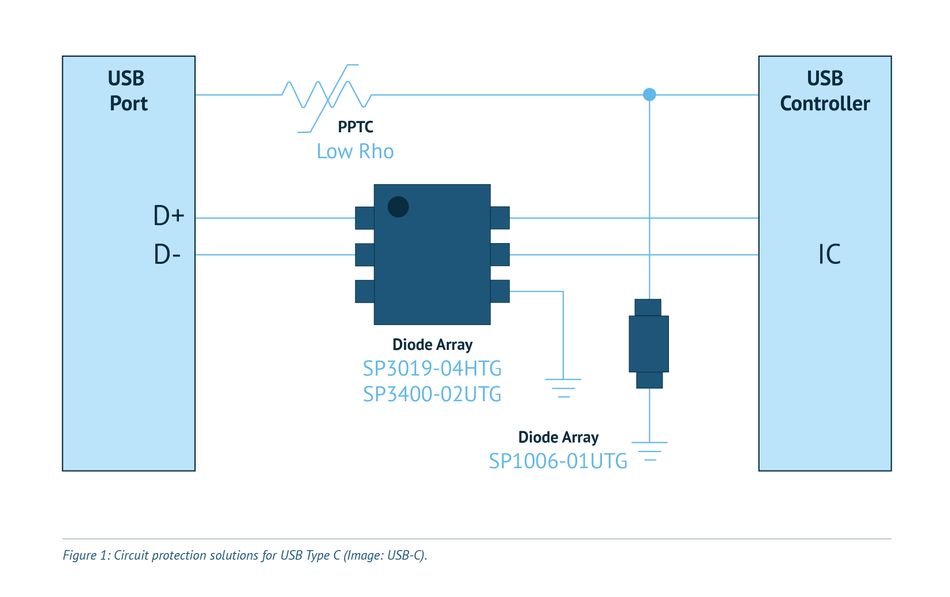
# 摘要
本文详细探讨了USB3.0 U盘技术的基础知识、设计要点、故障排查技术以及优化指南。首先介绍了
【MD290系列变频器安装与维护】:一步到位,确保操作无误且延长设备寿命(权威指南)
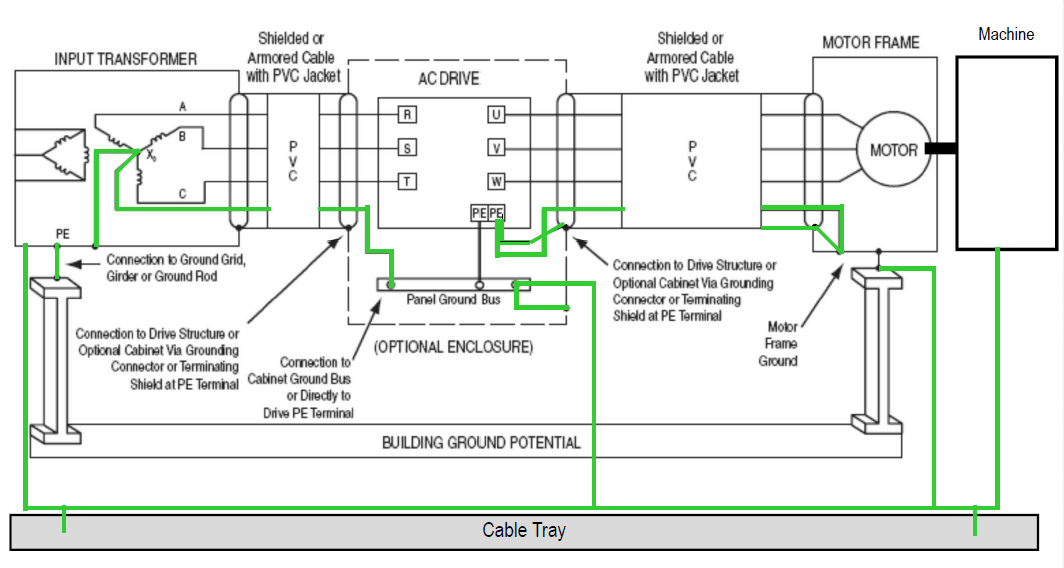
# 摘要
本文全面介绍了MD290系列变频器的基本使用、功能设定、维护保养及高级应用。首先概述了变频器的主要功能和技术参数,接着详细阐述了安装前的准备工作、安装步骤以及操作面板和软件配置方法。文章还重点讨论了维护保养的重要性和延长设备寿命的策略,以及如何通过特殊应用配置和系统集成提高变频器的性能。最后,
编程的艺术与情感:构建情感化应用的技术与设计思维深度剖析

# 摘要
随着技术的发展和用户需求的多样化,情感化应用越来越受到重视。本文首先定义了情感化应用的概念并强调了其在提升用户体验方面的重要性。继而,文章详细探讨了情感化设计的理论基础
【HFSS15启动故障快速解决指南】:20年专家教你如何诊断和修复启动问题(初学者必备)

# 摘要
本文详细探讨了HFSS15启动故障的原因、诊断技术和解决方法。首先,概述了HFSS15软件架构及启动流程,并分析了启动故障的类型及原因,包括常见错误代码、系统兼容性问题及第三方软件冲突。随后,深入介绍了诊断技术,包括日志文件分析、系统监控工具的使用和故障排除步骤。接着,提供了实践中的解决方法,涉及系统设置调整、常规故障处理和高级
【点云数据提取进阶】:深入解析ROS Bag点云信息提取的高级方法

# 摘要
本文深入探讨了ROS Bag数据结构及其在点云数据处理中的应用。文章首先介绍了ROS Bag文件格式和点云数据的理
关键性能指标(KPI)全面解析:中文版PACKML标准深度分析

# 摘要
PACKML标准作为一种用于包装机器的标准,其起源、发展及其在性能监测、分析与优化中的应用正逐渐受到关注。本文首先探讨了PACKML的起源和核心理念,包括机器生命周期模型、关键性能指标(KPI)的定义和标准操作模式。接着,文章深入分析了PACKML标准下的性能监测与分析技术要求,数据采集方法和实时监控系统搭建。文章还探讨了PACKML标准在自动化领域的应用,以及如
S3C2440A核心板时钟系统优化:原理图深度分析与实践指南

# 摘要
本文对S3C2440A核心板时钟系统进行了全面的分析与探讨,涵盖了时钟系统的基本原理、软件配置、优化实践以及进阶应用与未来展望。首先介绍了S3C2440A时钟源架构、时钟树和稳定性考量,包括晶振选择与电源噪声处理。接着,探讨了时钟系统软件配置方法、时钟管理策略以及调试和测试技巧。随后,
LMS算法完整指南:理论到实践,突破最小均方误差
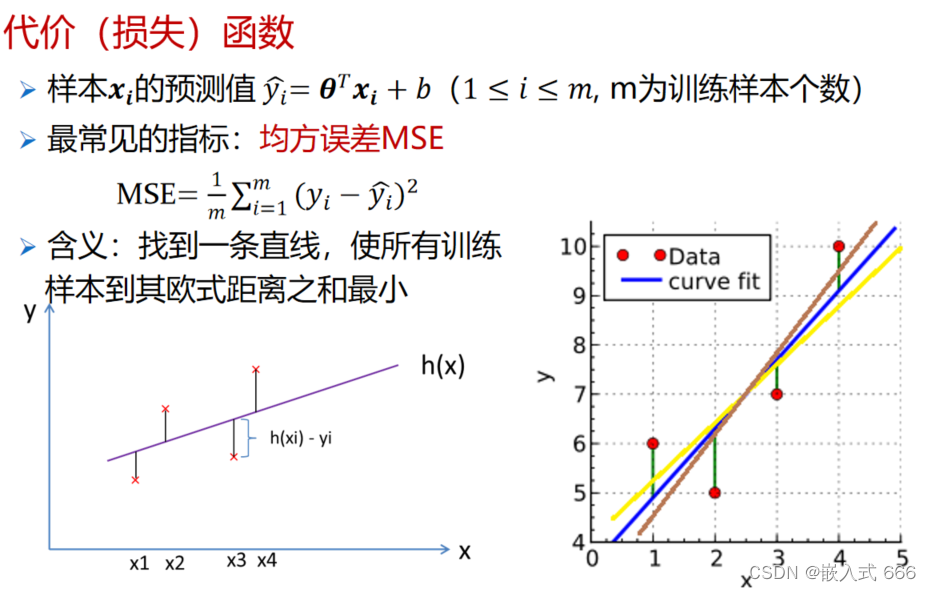
# 摘要
本文全面介绍了最小均方(LMS)算法的原理、应用场景、优化策略以及未来趋势。首先简要概述了LMS算法的基本概念及其在各种应用中的重要作用。其次,深入分析了LMS算法的理论基础,包括自适应滤波器的工作原理、算法的数学模型以及性能评估标准。随后,探讨了在实践中如何选择和调整LMS算法参数,通过MATLAB仿真和硬件实现(如FPGA和DSP处理器)来验证算法的有效性。文章还涉及了LMS算法的变种及其改进
提升加工精度:高级CNC技术应用策略揭秘
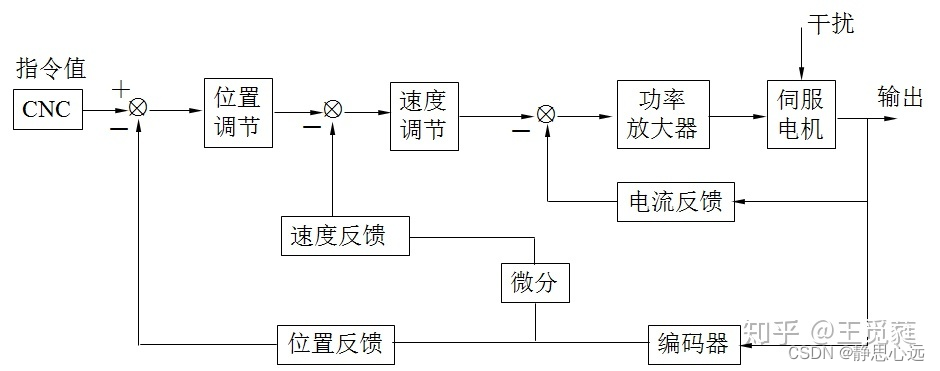
# 摘要
CNC技术作为一种高效率、高精度的机械加工方法,在现代制造业中占据核心地位。本文首先概述了CNC技术的基础知识、工作原理以及加工工艺流程,随后深入探讨了提高加工精度的关键技术和工艺优化方法。高级编程技巧章节分析了编程语言的应用、三维模型处理以及路径优化策略,同时介绍了调试与仿真技术在CNC编程中的重要性。接着,本文讨论了CNC系统与工业物联网的融合以及自动化解决方案在提高生产效率方面的作用。在展望CNC技术未来时,重点突出了新材料加工
极限的真谛:Apostol带你深入解析数学分析中的极限理论
# 摘要
极限是数学分析中的核心概念,为连续性、微分、积分等高级数学理论提供了基础。本文系统地探讨了极限的基本概念、严格定义,以及存在条件和性质,并深入分析了理论证明的技巧。通过介绍基本和复杂函数极限的计算方法,本文展示了极限在序列与级数中的应用。此外,本文还探讨了极限理论在数学分析其他领域的应用,包括连续性、微分学和积分学,并对极限理论在复分析和现代数学研究中的角色进行了讨论。文章最后对极限理论的学习方法提出了建议,并对当前研究动态和未来发展方向进行了展望。
# 关键字
极限;数学分析;ε-δ定义;序列与级数;微分学;积分学
参考资源链接:[Tom Apostol Mathematica
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )