【NHANES数据集】在生物统计学中的秘密武器:R包案例剖析
发布时间: 2024-12-29 11:32:41 阅读量: 17 订阅数: 19 

# 摘要
本文介绍了NHANES数据集及其在生物统计学中的应用背景,并深入探讨了R语言在处理此类数据集时的基础和高级功能。通过对NHANES数据集的结构、内容、清洗、预处理和高级处理技巧的详细分析,以及在生物统计分析中的具体应用案例,如描述性统计、推断统计、多变量分析、回归分析和预测建模技术的展示,本文意在说明R语言在生物统计学领域的重要作用和实践价值。同时,文中也展望了R语言在生物统计学领域未来的发展趋势以及社区在R包创新方面的贡献。
# 关键字
NHANES数据集;生物统计学;R语言;数据预处理;多变量统计分析;高级预测建模
参考资源链接:[使用nhanesR包进行数据提取的详细指南](https://wenku.csdn.net/doc/fabiisoq7m?spm=1055.2635.3001.10343)
# 1. NHANES数据集简介与应用背景
## 1.1 数据集概览
NHANES(National Health and Nutrition Examination Survey)是由美国疾病控制与预防中心(CDC)的国家卫生统计中心进行的一项连续调查。该数据集涉及广泛的人口健康指标,包括但不限于营养状况、健康行为、身体检查结果以及实验室测试数据。
## 1.2 应用背景的重要性
NHANES数据集提供了对美国公共健康状况的独特见解,尤其是在营养、肥胖、慢性病风险因素等领域。这些数据对于公共卫生政策制定者、医疗保健专业人员和研究人员而言,是极其宝贵的资源。它帮助我们理解复杂的人口健康模式,并通过数据分析来指导和评估公共健康干预措施。
## 1.3 数据集的实践意义
在实践中,NHANES数据集不仅被用来描述和解释健康与营养趋势,还被用于开发和校准健康风险评估工具。生物统计学家使用这些数据进行模型构建和验证,以预测疾病的发生,从而为个人和群体提供定制化的预防和干预方案。
通过介绍NHANES数据集,本文将引导读者深入理解其在生物统计学中的应用,并在后续章节中探讨如何利用R语言进行深入的数据分析。
# 2. R语言基础及其在生物统计学中的角色
## 2.1 R语言概述
### 2.1.1 R语言的历史和特点
R语言起源于20世纪90年代,由新西兰奥克兰大学的罗伯特·杰特曼(Robert Gentleman)和罗斯·伊哈卡(Ross Ihaka)开发。它是在S语言的基础上创建的,并且免费开源,因此得到了广泛的社区支持。R语言的特点可以概括为以下几点:
- **免费和开源**:R语言及其所有扩展包都是免费提供的,这意味着用户不必为获取软件或其更新支付费用。
- **强大的社区支持**:R拥有一个活跃的社区,为用户提供了大量的教程、代码片段、问题解答等资源。
- **丰富的统计和图形功能**:R语言内置了大量用于数据分析和可视化的功能,还支持通过CRAN(Comprehensive R Archive Network)安装第三方包以扩展其能力。
- **跨平台兼容性**:R可以在多种操作系统上运行,包括Windows、Mac OS X和Linux。
- **集成和自定义性**:R可以与其它语言(如C、C++、Fortran等)结合使用,也可以通过自定义函数进行扩展。
### 2.1.2 R语言在生物统计学中的重要性
在生物统计学领域,R语言的重要性日益凸显。以下是几个关键理由:
- **特定于领域的包**:生物统计学领域的研究人员开发了大量专门的R包来处理基因序列数据分析、生物信息学、流行病学等领域的问题。
- **数据可视化**:R语言中的ggplot2、lattice等包,提供了非常强大的数据可视化能力,使得研究者可以清晰地展示复杂的数据集。
- **统计建模**:R语言提供了一系列用于各种统计模型的包,包括线性回归、逻辑回归、生存分析等,这使得进行复杂统计分析成为可能。
- **共享和重复性**:数据分析和统计模型的脚本可以轻松地与同行共享,并且可以通过重复相同的代码步骤来验证结果。
## 2.2 R语言中的基础统计功能
### 2.2.1 数据类型和数据结构
R语言支持多种数据类型,包括数字、字符、逻辑值以及更复杂的数据结构,如向量、因子、列表和数据框(data.frame)。以下是R语言中一些基本的数据类型和结构的介绍:
- **向量(Vector)**:基本的数据结构,可以包含数值、字符或其他数据类型。
- **因子(Factor)**:用于表示分类数据的数据结构,每个因子值对应一个水平(level)。
- **列表(List)**:可以包含不同类型数据的复杂数据结构。
- **数据框(Data Frame)**:用于存储表格数据的数据结构,每一列可以是不同的数据类型。
### 2.2.2 基本的统计分析函数
R语言提供了一系列内置的统计函数,用于进行基础的描述性统计分析。这包括均值、中位数、标准差等,以及更高级的统计测试,例如t检验和ANOVA。下面是一些常用的基础统计函数:
```r
# 计算向量的均值
mean(x)
# 计算向量的中位数
median(x)
# 计算向量的标准差
sd(x)
# 执行t检验
t.test(x, y)
# 执行ANOVA
aov(y ~ x)
```
其中`x`和`y`是向量,`x`是自变量,`y`是因变量。这些函数使得R语言在进行快速的统计分析时非常方便。
### 2.2.3 R语言的数据可视化能力
R语言擅长数据可视化,可以创建各种静态图形,并可以生成动态图形。ggplot2是一个流行的R包,它提供了一种强大的方式来绘制图形。以下是一个使用ggplot2绘图的基本示例:
```r
library(ggplot2)
# 假设df是一个数据框,包含变量x和y
ggplot(df, aes(x, y)) +
geom_point() + # 添加点图层
geom_smooth() # 添加平滑曲线
```
该代码使用数据框`df`中的变量`x`和`y`创建一个散点图,并添加一条平滑曲线来表示数据的趋势。
## 2.3 R语言的高级特性
### 2.3.1 包管理与CRAN
R的包管理通过一系列的函数实现,最常用的有`install.packages()`和`library()`。CRAN是R软件包的主要存储库,它使得用户可以方便地安装和更新软件包。
### 2.3.2 与其他语言的接口
R可以与多种其他编程语言进行接口,包括C、C++、Python等。Rcpp包使得在R中集成C++代码变得简单,而Rserve和rpy2等包允许R与其他语言进行通信。
### 2.3.3 R语言的并行计算和性能优化
R语言通过并行包(如parallel包)和一些特定任务的包,比如foreach和doParallel,支持并行计算。性能优化可以在编写更高效的R代码的基础上,通过并行计算进一步提高。
以上为第二章中关于R语言基础及其在生物统计学中角色的详细内容。下面将继续展开下一章节内容。
# 3. NHANES数据集探索与预处理
## 3.1 NHANES数据集的结构和内容
NHANES(National Health and Nutrition Examination Survey)数据集是美国国家卫生统计中心发布的公开数据集,涵盖了美国民众的健康和营养状况的广泛信息。本小节将深入探讨数据集的获取和读取方法,以及如何对数据集进行初步的描述性分析。
### 3.1.1 数据集的获取和读取
NHANES数据集可以通过多个途径获取,最直接的方式是访问美国国家健康统计中心的官方网站。数据通常以SAS格式发布,但也可以通过特定工具转换为R语言可以直接读取的格式。
在R中,可以使用`haven`包来读取SAS格式的数据文件:
```r
# 安装并加载haven包
install.packages("haven")
library(haven)
# 读取SAS格式的NHANES数据
nhanes_data <- read_sas("path/to/NHANES_data.sas7bdat")
# 查看数据结构
str(nhanes_data)
```
上述代码块展示了如何安装和加载`haven`包,以及如何使用`read_sas()`函数读取SAS格式的数据。`str()`函数用于输出数据的结构信息,有助于我们了解数据集的框架和内容。
### 3.1.2 数据集的基本描述性分析
在获取数据集之后,通常需要进行一些基础的描述性分析,以便了解数据集中包含的变量和数据的基本特征。描述性分析可能包括计算均值、中位数、最小值、最大值、标准差等统计量。
```r
# 计算数值变量的描述性统计量
summary(nhanes_data$height) # 以身高为例
summary(nhanes_data$weight)
# 查看分类变量的频数分布
table(nhanes_data$gender)
```
在上述代码中,`summary()`函数用于得到数值变量的高度和体重的描述性统计量,而`table()`函数用于展示分类变量(例如性别)的频数分布。这些基础统计量为我们提供对数据集的初步了解。
## 3.2 数据清洗与预处理技巧
数据预处理是数据分析过程中非常重要的一个环节。在这一节中,我们将学习如何处理缺失值、异常值,并进行数据转换和归一化。
### 3.2.1 缺失值的处理
在现实世界的数据集中,缺失值是常见的问题。R语言提供了多种方法来处理这些缺失值。
```r
# 查看含有缺失值的变量
any(is.na(nhanes_data$age))
# 删除含有缺失值的行
nhanes_data_complete <- na.omit(nhanes_data)
# 使用平均值填充缺失值
nhanes_data_mean <- transform(nhanes_data, age = ifelse(is.na(age), mean(age, na.rm = TRUE), age))
```
代码块解释了三种常见的处理缺失值的方法:检查含有缺失值的变量、删除含有缺失值的行以及使用平均值填充缺失值。`na.omit()`函数用于删除含有缺失值的数据行,而`transform()`和`ifelse()`组合用于条件性填充缺失值。
### 3.2.2 异常值的识别与处理
异常值是指那些与数据集中其他观测值显著不同的值。R中可以通过箱线图来识别异常值。
```r
# 绘制年龄的箱线图以识别异常值
boxplot(nhanes_data$age, main = "Boxplot of Age")
# 删除年龄中的异常值
age_no_outliers <- nhanes_data$age[abs(nhanes_data$age - median(nhanes_data$age, na.rm = TRUE)) <= 1.5 * IQR(nhanes_data$age, na.rm = TRUE)]
```
通过绘制年龄的箱线图,可以识别出异常值。随后使用IQR(四分位距)方法识别并移除异常值。这里展示了一个有效识别和处理异常值的案例。
### 3.2.3 数据转换和归一化
数据转换和归一化是调整数据分布,使其更好地符合某些分析方法要求的过程。常见的数据转换方法包括对数转换、平方根转换等,而归一化通常指将数据缩放到[0, 1]区间。
```r
# 对数转换示例
log_age <- log(nhanes_data$age)
# 归一化示例
normalized_weight <- (nhanes_data$weight - min(nhanes_data$weight)) / (max(nhanes_data$weight) - min(nhanes_data$weight))
```
这里,`log()`函数用于对年龄变量进行对数转换,而归一化处理使用了最大最小值法。这两种转换技术对于改善数据分析结果非常有用。
## 3.3 数据集的高级处理
在数据集经过基础的预处理后,我们需要应用一些高级技术对数据进行进一步的处理。
### 3.3.1 特征提取和选择
特征提取和选择可以减少模型的复杂度,提高模型的准确率。对于NHANES数据集,特征选择可能包括基于统计测试的选择方法和基于模型的选择方法。
```r
# 使用基于统计测试的选择方法
library(caret)
control <- rfeControl(functions=rfFuncs, method="cv", number=10)
results <- rfe(nhanes_data[,-1], nhanes_data[,1], rfeControl=control)
# 使用基于模型的选择方法
library("leaps")
regfit.full <- regsubsets(SBP ~ ., data=nhanes_data, nvmax=20)
summary(regfit.full)
```
`rfe()`函数来自`caret`包,用于递归特征消除,而`regsubsets()`函数来自`leaps`包,用于对线性模型的不同子集进行评估,选择最佳特征组合。
### 3.3.2 数据集的分割与重采样
数据集分割是将数据集分割成训练集和测试集,这对于模型的训练和验证至关重要。重采样技术如交叉验证可以用来估计模型的性能。
```r
# 数据集分割
set.seed(123) # 确保结果可复现
split_index <- createDataPartition(nhanes_data$gender, p = 0.8, list = FALSE)
train_data <- nhanes_data[split_index,]
test_data <- nhanes_data[-split_index,]
# 重采样:k折交叉验证
control <- trainControl(method="cv", number=10)
model <- train(SBP ~ ., data=train_data, method="lm", trControl=control)
```
上述代码块展示了如何使用`createDataPartition()`函数创建训练和测试数据集,并应用`trainControl()`和`train()`函数来进行10折交叉验证。这些步骤是数据科学项目中常见的实践。
在本小节中,我们详细介绍了NHANES数据集的获取、读取和基础描述性分析方法,深入探讨了数据清洗、预处理技巧和高级处理技术。这些内容为后续的生物统计分析奠定了坚实的基础,为科研人员和数据分析专业人士提供了一套完整的数据分析流程。在下一章中,我们将利用R包进行生物统计分析,进一步探索NHANES数据集的统计学特征。
# 4. 使用R包进行生物统计分析
## 4.1 NHANES数据集的描述性统计分析
在生物统计学的研究中,描述性统计分析是理解数据集基本特征的基础。NHANES数据集通过提供详尽的健康和营养信息,成为生物统计分析中的宝贵资源。这一节将介绍如何使用R语言及其包来实现NHANES数据集的描述性统计分析。
### 4.1.1 分组统计与汇总报告
分组统计是按不同变量对数据集进行分类并计算每个类别的汇总统计数据。在R中,我们可以使用`dplyr`包来高效地进行此类操作。以下是一个示例代码:
```r
library(dplyr)
# NHANES数据集的分组统计示例
grouped_stats <- NHANES_adult %>%
group_by(Gender) %>%
summarise(
Average_Age = mean(Age, na.rm = TRUE),
Total_Population = n(),
Average_BMI = mean(BMI, na.rm = TRUE)
)
```
在这段代码中,我们首先加载了`dplyr`包,然后使用管道操作符(%>%)将NHANES_adult数据集传递到一系列操作中。我们按性别(Gender)分组,并计算每个性别组中的平均年龄(Average_Age)、总人口数(Total_Population)以及平均BMI(Average_BMI)。参数`na.rm = TRUE`用于忽略缺失值(NA),确保计算平均值时不会因为NA值导致结果失真。
### 4.1.2 数据分布的图形化展示
数据分布的图形化展示有助于直观理解数据特征,是描述性统计分析中的一个重要环节。R语言的`ggplot2`包是一个功能强大的绘图工具,可以创建各种类型的统计图形。以下是一个绘制NHANES数据集中BMI分布的直方图的代码示例:
```r
library(ggplot2)
# NHANES数据集中BMI分布的直方图
ggplot(NHANES_adult, aes(x = BMI)) +
geom_histogram(binwidth = 1, fill = "skyblue", color = "black") +
labs(title = "BMI Distribution in NHANES Dataset",
x = "BMI",
y = "Frequency") +
theme_minimal()
```
在此代码段中,我们使用`ggplot`函数初始化绘图,并指定数据集(NHANES_adult)和数据点映射(BMI)。`geom_histogram`函数用于生成直方图,`binwidth`参数定义了每个柱状图的宽度。图表的标题和坐标轴标签通过`labs`函数添加,`theme_minimal`函数用来设置一个简洁的主题样式。
## 4.2 推断统计分析的实现
推断统计分析允许研究者基于样本数据对总体参数进行推断,是生物统计学中的核心内容。本节将介绍如何使用R语言实现假设检验和置信区间的计算。
### 4.2.1 假设检验
假设检验是推断统计中的一个重要工具,用于评估统计结果是否具有统计学意义。在R中,`t.test`函数提供了进行t检验的能力,而`chisq.test`函数则用于卡方检验。以下是使用`t.test`函数进行两样本t检验的一个示例:
```r
# NHANES数据集的两样本t检验示例
t_test_result <- t.test(
NHANES_adult$Weight[NHANES_adult$Gender == "male"],
NHANES_adult$Weight[NHANES_adult$Gender == "female"]
)
print(t_test_result)
```
在这段代码中,我们对NHANES_adult数据集中的男性和女性体重(Weight)进行了两样本t检验。结果通过`print`函数输出,包括t值、自由度(df)、p值以及总体均值差的95%置信区间等。
### 4.2.2 置信区间和效应量计算
置信区间和效应量是衡量统计结果重要性的关键指标。在R中,许多统计函数都直接返回置信区间。效应量通常需要额外的计算。以下是计算均值差异的效应量的一个示例:
```r
# 计算均值差异的效应量
mean_diff <- mean(NHANES_adult$Weight[NHANES_adult$Gender == "male"]) -
mean(NHANES_adult$Weight[NHANES_adult$Gender == "female"])
# 标准误差计算
se_diff <- sqrt(var(NHANES_adult$Weight[NHANES_adult$Gender == "male"]) /
sum(NHANES_adult$Gender == "male") +
var(NHANES_adult$Weight[NHANES_adult$Gender == "female"]) /
sum(NHANES_adult$Gender == "female"))
# 计算效应量
cohen_d <- mean_diff / se_diff
cat("Cohen's d:", cohen_d)
```
在这段代码中,我们首先计算了男性和女性体重的均值差异。然后计算了两个群体体重的标准误差(se_diff),最终使用Cohen的d公式计算了效应量(cohen_d),这是衡量两组均值差异的标准化指标。
## 4.3 多变量统计分析方法
多变量统计分析用于研究同时涉及多个变量的情况,本节将介绍如何使用R进行主成分分析、因子分析、聚类分析以及生存分析和时间序列分析。
### 4.3.1 主成分分析(PCA)
主成分分析(PCA)是一种降维技术,可以用于减少数据集中的变量数量,同时尽可能保留原始数据的信息。以下是一个使用`prcomp`函数进行PCA的代码示例:
```r
# NHANES数据集的PCA分析示例
pca_result <- prcomp(NHANES_adult[, c("Age", "Weight", "Height", "BMI")], scale. = TRUE)
# 绘制PCA的前两个主成分的散点图
plot(pca_result$x[, 1:2], col = NHANES_adult$Gender, pch = 19)
legend("topright", legend = levels(NHANES_adult$Gender), col = 1:length(levels(NHANES_adult$Gender)), pch = 19)
```
在这段代码中,我们使用`prcomp`函数对年龄(Age)、体重(Weight)、身高(Height)和BMI四个变量进行PCA,并设置`scale.= TRUE`以标准化数据。`plot`函数绘制了前两个主成分的散点图,并按性别着色。`legend`函数用于添加图例。
### 4.3.2 因子分析和聚类分析
因子分析和聚类分析是探索数据结构和发现隐藏模式的有效方法。在R中,`factanal`函数可以执行因子分析,而`kmeans`函数是进行聚类分析的常用工具。以下是聚类分析的一个示例:
```r
# 对NHANES数据集进行K均值聚类分析
set.seed(123) # 确保结果可复现
kmeans_result <- kmeans(NHANES_adult[, c("Age", "Weight", "Height", "BMI")], centers = 3)
# 将聚类结果添加到原始数据中
NHANES_adult$Cluster <- kmeans_result$cluster
# 绘制聚类结果
clusplot(NHANES_adult[, c("Age", "Weight", "Height", "BMI")], kmeans_result$cluster, color = TRUE, shade = TRUE, labels = 2, lines = 0)
```
在这段代码中,我们首先使用`set.seed`函数确保聚类结果的可复现性。然后,`kmeans`函数对年龄、体重、身高和BMI进行聚类,设定3个聚类中心。聚类结果通过`CLUSPLOT`函数绘制成散点图,展示每个聚类的分布情况。
### 4.3.3 生存分析和时间序列分析
生存分析和时间序列分析用于分析特定类型的数据,例如生存时间或时间序列数据。在R中,`survival`包提供了生存分析的功能,而`ts`类和相关函数用于时间序列分析。
生存分析的一个典型应用是对疾病生存时间的分析。以下是一个使用`survfit`函数进行生存曲线绘制的示例:
```r
# 生存分析示例代码
# 注意:此处仅为示例,真实的生存分析需要完整的生存时间和事件发生的样本数据
surv_obj <- Surv(time = NHANES_adult$Survival_Time, event = NHANES_adult$Event)
# 使用kaplan-meier方法绘制生存曲线
km_fit <- survfit(surv_obj ~ 1)
plot(km_fit)
```
在时间序列分析中,R可以处理和分析时间点序列数据。以下是一个时间序列分析的简单示例:
```r
# 时间序列分析示例代码
# 假设NHANES_adult有一个时间序列变量Time_Series
ts_data <- ts(NHANES_adult$Time_Series, start = c(1999, 1), frequency = 12)
# 绘制时间序列图
plot(ts_data)
# 应用时间序列分解
decompose_ts <- decompose(ts_data, "additive")
plot(decompose_ts)
```
在上述代码中,我们首先将NHANES_adult数据集中的一个时间序列变量转换为时间序列对象`ts_data`,然后使用`plot`函数绘制时间序列图。接着,我们使用`decompose`函数将时间序列分解为趋势、季节性和随机成分,并绘制分解结果图。
# 5. NHANES数据集的高级分析案例
## 5.1 NHANES数据集中的回归分析
### 5.1.1 线性回归和逻辑回归
线性回归模型是统计学中非常基础且广泛使用的预测模型。在生物统计学中,线性回归可以用于分析一个或多个连续变量之间的关系。例如,我们可以通过NHANES数据集来探究个人的胆固醇水平与其生活习惯(如饮食和运动)之间的相关性。
```R
# 加载所需的R包
library(nh4e)
# 假设我们使用NHANES数据集中的'age', 'bmi', 'chol'变量
# 导入NHANES数据集
data(NHANES)
# 创建线性模型,以'chol'作为响应变量,'age'和'bmi'作为解释变量
lm_model <- lm(chol ~ age + bmi, data = NHANES)
# 输出模型摘要以查看回归分析结果
summary(lm_model)
```
在上述代码块中,我们首先加载了`nh4e`包,这可能是一个自定义的包,或者是示例中虚构的包,用于处理NHANES数据集。然后我们导入数据集,并构建一个线性回归模型,其中`lm()`函数用于拟合线性模型。模型摘要通过`summary()`函数输出,以查看模型的统计显著性、系数估计以及模型拟合优度等重要统计量。
在生物统计学中,逻辑回归通常用于处理因变量为二分类的情况。例如,若我们要分析是否患有高血压与年龄、体重指数(BMI)和性别之间的关系,我们可以采用逻辑回归模型。
```R
# 创建逻辑回归模型,以'hypertension'作为响应变量(二分类变量),'age', 'bmi', 'gender'作为解释变量
logit_model <- glm(hypertension ~ age + bmi + gender, data = NHANES, family = binomial)
# 输出模型摘要
summary(logit_model)
```
这里我们使用`glm()`函数来进行逻辑回归,注意`family`参数被设定为`binomial`,表明我们使用的是二项分布作为链接函数。模型摘要同样提供了我们分析所需的关键统计信息。
### 5.1.2 回归诊断和模型优化
在拟合回归模型后,一个重要的步骤是对模型的诊断,以确保模型的假设是满足的,并检查是否有潜在的问题,如异方差性、多重共线性、异常值或者影响点。R语言中提供了许多用于回归诊断的工具和方法。
```R
# 检查残差图
par(mfrow=c(2,2))
plot(lm_model)
par(mfrow=c(1,1))
# 检查多重共线性
vif(lm_model)
# 查找影响点和杠杆点
influence.measures(lm_model)
```
在这些诊断步骤中,我们首先通过绘制残差图来检查线性模型的假设,其中包括残差与拟合值的图、标准化残差的直方图、标准化残差的正态QQ图以及杠杆值图等。然后,我们使用`vif()`函数来检查多重共线性,最后使用`influence.measures()`函数来识别可能的高杠杆点和影响点。
模型优化可能包括添加或删除变量、变换变量、处理缺失数据、或者使用更复杂的模型来提高预测的准确度。通过模型优化,我们可以使模型更好地适应数据,减少偏差和方差,从而提高其在新数据上的泛化能力。
## 5.2 高级预测建模技术
### 5.2.1 机器学习在生物统计中的应用
在生物统计学领域,机器学习算法如随机森林、支持向量机和深度学习正被越来越多地用于复杂数据集的预测建模和模式识别。这些技术能够处理大规模的数据集,识别非线性关系,并处理高维数据。
以随机森林算法为例,它可以用于处理NHANES数据集中的分类问题,例如预测个人是否患有某种慢性疾病。
```R
# 使用randomForest包来拟合随机森林模型
library(randomForest)
# 创建随机森林模型
rf_model <- randomForest(hypertension ~ ., data = NHANES)
# 输出模型性能
print(rf_model)
```
上述代码展示了如何利用`randomForest`包来创建一个随机森林模型,并预测`hypertension`。我们使用了`hypertension`作为响应变量,而`.`表示模型中使用了数据集的所有其他变量作为解释变量。`print()`函数用于输出模型的基本性能指标。
### 5.2.2 模型的选择、评估与验证
在建模过程中,选择正确的模型是至关重要的。需要根据数据的特性和研究问题来选择合适的模型。一旦模型被选定,就需要对其进行评估和验证,以确定其预测能力。
评估模型通常需要使用交叉验证,这是一种统计方法,用于评估并比较学习算法的泛化能力。我们可以使用不同的评估指标来衡量模型的性能,如准确度、精确度、召回率和AUC值。
```R
# 使用caret包进行模型评估
library(caret)
# 设置交叉验证的参数
train_control <- trainControl(method = "cv", number = 10)
# 训练一个逻辑回归模型,并使用交叉验证
logit_model_cv <- train(hypertension ~ ., data = NHANES, method = "glm", family = "binomial", trControl = train_control)
# 输出交叉验证的结果
print(logit_model_cv)
```
在上述代码中,我们使用了`caret`包来设置交叉验证,并使用`train()`函数来拟合逻辑回归模型,并应用了10折交叉验证。最终我们输出了交叉验证的结果,这有助于我们了解模型在未知数据上的预测性能。
通过这些高级预测建模技术和模型评估方法,研究者可以在生物统计学中进行更为深入和复杂的分析,从而得到更可靠的研究结论。
# 6. R在生物统计学中的未来展望
## 6.1 生物统计学的发展趋势与挑战
随着科技的不断进步,生物统计学作为一门应用广泛的学科,正面临着前所未有的发展机遇与挑战。其中,大数据技术的兴起为生物统计学的发展提供了巨大的推动力,与此同时,传统的统计软件与编程语言,如R语言,也在积极适应这一趋势,以满足日益复杂的数据处理需求。
### 6.1.1 大数据在生物统计中的作用
大数据技术的应用,使得我们能够处理以前无法想象规模的数据集,这对于生物统计学来说意味着更全面的分析和更深的洞察力。在临床试验、基因组学、药物研发等领域,大数据带来的结果往往是革命性的。例如,在基因组学研究中,大数据技术可以帮助科学家们快速分析基因序列,识别疾病相关的遗传标记。
### 6.1.2 R语言的持续发展和改进
R语言作为生物统计学领域的一个重要工具,它正在不断地进行更新和改进以适应大数据时代的需求。包括加强其对并行计算的支持,引入更高效的数据处理和分析方法,以及不断优化和升级其图形用户界面。R语言社区也在积极贡献新的包,这些新包往往专注于特定的统计方法或数据分析技术,极大地扩展了R语言在生物统计中的应用范围和深度。
## 6.2 R包的创新与未来应用
R语言的包管理器CRAN是该语言生态系统的核心。它让R语言拥有了几乎可以完成任何统计分析任务的包。CRAN上不断涌现的新包反映了R语言社区的活跃度以及对创新的追求。
### 6.2.1 新兴R包和工具的探索
为了应对大数据的挑战,新的R包不断被开发出来,例如,用于处理大规模数据集的`data.table`包,以及用于高性能计算的`foreach`包和`parallel`包。此外,针对特定生物统计问题,如生存分析和遗传数据处理,也有专用的包如`survival`和`SNPRelate`提供解决方案。R社区也在推动R语言与其他编程语言的交流与融合,例如R与Python的互操作性包`reticulate`,为用户提供了更多的灵活性和选择。
### 6.2.2 生物统计学研究中的R语言社区贡献
R语言社区是由全球的科学家和统计学家组成的庞大网络,他们通过贡献代码、提供教程和举办研讨会等方式,不断推动R语言的发展。社区不仅提供技术上的支持,更是生物统计学创新的孵化器。社区成员经常合作解决复杂的统计问题,分享他们的知识和经验,这对于生物统计学的进步至关重要。
在R语言和生物统计学的未来展望中,我们将看到更多的自动化分析工具和用户友好的接口的出现,这将极大地降低统计分析的门槛,使得非专业统计学家也能参与到复杂的数据分析中来。同时,R语言将继续以其开放性和灵活性,成为生物统计学研究中不可或缺的一部分。

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
“NHANES R 包学习笔记”专栏是一份全面的指南,涵盖了使用 NHANES R 包进行数据分析的各个方面。从新手入门到高级分析,该专栏提供了逐步的指导,涵盖了数据预处理、探索性分析、生存分析、时间序列分析、复杂样本设计数据分析、数据可视化、临床研究应用、编程技巧和探索性数据分析的高级策略。该专栏旨在帮助研究人员、数据科学家和公共卫生专业人员充分利用 NHANES 数据集,进行深入的数据分析和洞察力。
专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
潮流分析的艺术:PSD-BPA软件高级功能深度介绍

# 摘要
电力系统分析在保证电网安全稳定运行中起着至关重要的作用。本文首先介绍了潮流分析的基础知识以及PSD-BPA软件的概况。接着详细阐述了PSD-BPA的潮流计算功能,包括电力系统的基本模型、潮流计算的数学原理以及如何设置潮流计算参数。本文还深入探讨了PSD-BPA的高级功
RTC4版本迭代秘籍:平滑升级与维护的最佳实践

# 摘要
本文重点讨论了RTC4版本迭代的平滑升级过程,包括理论基础、实践中的迭代与维护,以及维护与技术支持。文章首先概述了RTC4的版本迭代概览,然后详细分析了平滑升级的理论基础,包括架构与组件分析、升级策略与计划制定、技术要点。在实践章节中,本文探讨了版本控制与代码审查、单元测试
SSD1306在智能穿戴设备中的应用:设计与实现终极指南
# 摘要
SSD1306是一款广泛应用于智能穿戴设备的OLED显示屏,具有独特的技术参数和功能优势。本文首先介绍了SSD1306的技术概览及其在智能穿戴设备中的应用,然后深入探讨了其编程与控制技术,包括基本编程、动画与图形显示以及高级交互功能的实现。接着,本文着重分析了SSD1306在智能穿戴应用中的设计原则和能效管理策略,以及实际应用中的案例分析。最后,文章对SSD1306未来的发展方向进行了展望,包括新型显示技术的对比、市场分析以及持续开发的可能性。
# 关键字
SSD1306;OLED显示;智能穿戴;编程与控制;用户界面设计;能效管理;市场分析
参考资源链接:[SSD1306 OLE
PM813S内存管理优化技巧:提升系统性能的关键步骤,专家分享!
# 摘要
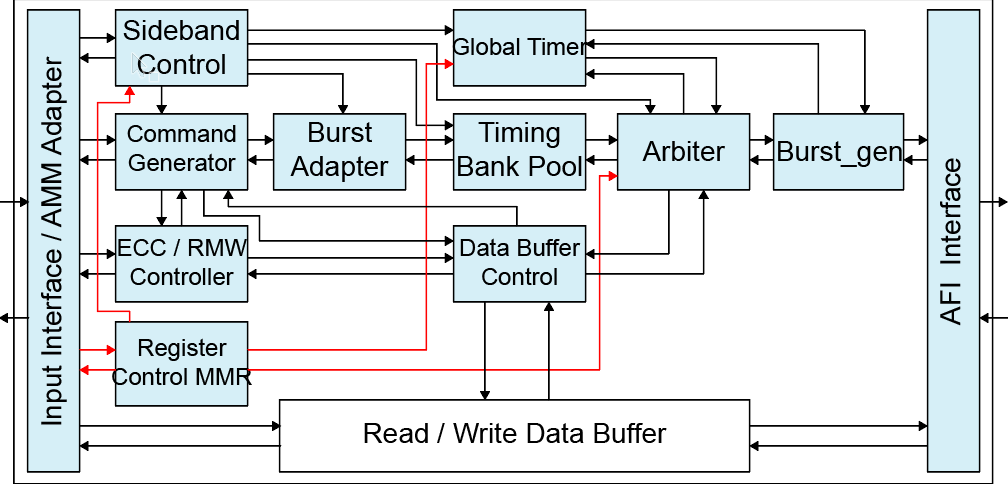
PM813S作为一款具有先进内存管理功能的系统,其内存管理机制对于系统性能和稳定性至关重要。本文首先概述了PM813S内存管理的基础架构,然后分析了内存分配与回收机制、内存碎片化问题以及物理与虚拟内存的概念。特别关注了多级页表机制以及内存优化实践技巧,如缓存优化和内存压缩技术的应用。通过性能评估指标和调优实践的探讨,本文还为系统监控和内存性能提
嵌入式系统中的BMP应用挑战:格式适配与性能优化
# 摘要
本文综合探讨了BMP格式在嵌入式系统中的应用,以及如何优化相关图像处理与系统性能。文章首先概述了嵌入式系统与BMP格式的基本概念,并深入分析了BMP格式在嵌入式系统中的应用细节,包括结构解析、适配问题以及优化存储资源的策略。接着,本文着重介绍了BMP图像的处理方法,如压缩技术、渲染技术以及资源和性能优化措施。最后,通过具体应用案例和实践,展示了如何在嵌入式设备中有效利用BMP图像,并探讨了开发工具链的重要性。文章展望了高级图像处理技术和新兴格式的兼容性,以及未来嵌入式系统与人工智能结合的可能方向。
# 关键字
嵌入式系统;BMP格式;图像处理;性能优化;资源适配;人工智能
参考资
【光辐射测量教育】:IT专业人员的培训课程与教育指南

# 摘要
光辐射测量是现代科技中应用广泛的领域,涉及到基础理论、测量设备、技术应用、教育课程设计等多个方面。本文首先介绍了光辐射测量的基础知识,然后详细探讨了不同类型的光辐射测量设备及其工作原理和分类选择。接着,本文分析了光辐射测量技术及其在环境监测、农业和医疗等不同领域的应用实例。教育课程设计章节则着重于如何构建理论与实践相结合的教育内容,并提出了评估与反馈机制。最后,本文展望了光辐射测量教育的未来趋势,讨论了技术发展对教育内容和教
【Ubuntu 16.04系统更新与维护】:保持系统最新状态的策略
# 摘要
本文针对Ubuntu 16.04系统更新与维护进行了全面的概述,探讨了系统更新的基础理论、实践技巧以及在更新过程中可能遇到的常见问题。文章详细介绍了安全加固与维护的策略,包括安全更新与补丁管理、系统加固实践技巧及监控与日志分析。在备份与灾难恢复方面,本文阐述了
ECOTALK数据科学应用:机器学习模型在预测分析中的真实案例

# 摘要
本文对机器学习模型的基础理论与技术进行了综合概述,并详细探讨了数据准备、预处理技巧、模型构建与优化方法,以及预测分析案例研究。文章首先回顾了机器学习的基本概念和技术要点,然后重点介绍了数据清洗、特征工程、数据集划分以及交叉验证等关键环节。接
分析准确性提升之道:谢菲尔德工具箱参数优化攻略

# 摘要
本文介绍了谢菲尔德工具箱的基本概念及其在各种应用领域的重要性。文章首先阐述了参数优化的基础理论,包括定义、目标、方法论以及常见算法,并对确定性与随机性方法、单目标与多目标优化进行了讨论。接着,本文详细说明了谢菲尔德工具箱的安装与配置过程,包括环境选择、参数配置、优化流程设置以及调试与问题排查。此外,通过实战演练章节,文章分析了案例应用,并对参数调优的实验过程与结果评估给出了具体指
CC-LINK远程IO模块AJ65SBTB1现场应用指南:常见问题快速解决
# 摘要
CC-LINK远程IO模块作为一种工业通信技术,为自动化和控制系统提供了高效的数据交换和设备管理能力。本文首先概述了CC-LINK远程IO模块的基础知识,接着详细介绍了其安装与配置流程,包括硬件的物理连接和系统集成要求,以及软件的参数设置与优化。为应对潜在的故障问题,本文还提供了故障诊断与排除的方法,并探讨了故障解决的实践案例。在高级应用方面,文中讲述了如何进行编程与控制,以及如何实现系统扩展与集成。最后,本文强调了CC-LINK远程IO模块的维护与管理的重要性,并对未来技术发展趋势进行了展望。
# 关键字
CC-LINK远程IO模块;系统集成;故障诊断;性能优化;编程与控制;维护
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )