Avogadro实战指南:分子编辑与构建一步到位
发布时间: 2024-12-03 23:29:59 阅读量: 71 订阅数: 37 

Avogadro:直观的分子编辑器和可视化工具-开源
参考资源链接:[Avogadro中文教程:分子建模与可视化全面指南](https://wenku.csdn.net/doc/6b8oycfkbf?spm=1055.2635.3001.10343)
# 1. Avogadro软件概述与安装
## 1.1 Avogadro软件简介
Avogadro是一款免费开源的分子编辑和可视化软件,广泛应用于化学、材料科学、分子生物学等领域。它支持多种操作系统,包括Windows、Linux和Mac OS。Avogadro以其直观的用户界面和强大的功能,为研究者提供了一个平台,可以方便地构建和编辑分子模型,进行分子几何优化,以及执行基本的量子化学计算。
## 1.2 安装Avogadro
要安装Avogadro,您可以访问其官方网站或GitHub页面下载适合您操作系统的安装程序。安装过程简单,通常只需要点击下一步进行默认安装即可。在安装过程中,您可以选择安装额外的分子数据文件和插件,以增加软件的功能。
```bash
# Windows 示例安装命令(以管理员权限运行)
Avogadro-1.2.0-win64.exe
```
安装完成后,启动Avogadro,您会看到一个简洁的用户界面,它包含了用于创建和编辑分子的主要工具。
## 1.3 Avogadro的功能特点
Avogadro提供了丰富的功能,包括但不限于:
- 分子建模:从基础元素到复杂分子结构的创建。
- 分子编辑:添加、移除原子和化学键,或修改分子结构。
- 可视化工具:提供了多种渲染选项,如球棒模型、线框模型、空间填充模型等,以更好地展示分子结构。
- 导入导出功能:支持多种化学文件格式,便于与其它化学软件数据交换。
以上内容仅为第一章的内容概要,后续章节将进一步深入探讨Avogadro的使用方法和在化学研究中的实际应用。
# 2. 分子建模基础理论
### 2.1 分子建模的重要性
分子建模是化学研究的一个重要领域,它将化学理论、数学计算和计算机技术相结合,对分子结构进行模拟。它的重要性不仅体现在基础研究中,而且在材料科学、药物设计、生物工程等众多领域都发挥着关键作用。
#### 2.1.1 理解分子建模在化学中的作用
分子建模能够帮助科学家预测分子间的相互作用,理解化学反应的机理,设计新的分子结构。随着计算能力的增强和算法的改进,分子建模已经成为化学实验的有效补充,可以在不需要实际合成和实验的情况下,预测分子的性质和反应性。
```mermaid
graph TD
A[分子建模] -->|辅助| B[化学实验]
A -->|增强理解| C[化学反应机理]
A -->|设计新分子| D[新药物分子]
A -->|预测性质| E[材料性质]
```
通过分子建模,化学家能够更深入地理解分子结构与功能之间的关系,从而在药物开发、材料设计等方面发挥巨大的潜力。例如,在药物设计中,分子建模可以帮助研究者筛选出最有潜力的候选分子,从而减少实验成本和时间。
#### 2.1.2 分子建模的科学基础
分子建模基于量子力学和统计力学的基本原理。它通常涉及到分子力学、分子动力学以及量子化学计算等方法。这些方法可以模拟分子系统的微观行为,从而在原子级别上理解化学和物理现象。
### 2.2 分子几何与化学键
分子几何指的是分子中各原子的三维空间排布,而化学键则是原子间相互作用形成的稳定连接。分子几何和化学键是描述分子结构的两个基本要素。
#### 2.2.1 几何学在分子建模中的应用
在分子建模中,几何学的应用至关重要。分子的几何结构决定了其物理和化学性质。比如,碳原子的sp3、sp2和sp杂化状态影响了碳化合物的反应性。分子建模软件通常提供各种几何优化工具,帮助用户根据最小能量原理精确地确定分子的稳定构型。
几何优化的算法通常涉及到力场的计算,计算出每个原子所受的力,并通过迭代的方式找到能量最低的稳定状态。此过程涉及到复杂的数学运算,现代分子建模软件如Avogadro能够自动处理这些计算。
#### 2.2.2 化学键的类型及其表示方法
化学键可以分为共价键、离子键、金属键等多种类型,每种键在分子建模中都有其特定的表示方法。在模拟过程中,这些键的信息需要准确地输入到模型中。
共价键通常用键长和键角来表示,而离子键和金属键则需要考虑电荷分布和电荷转移。为了更精确地模拟键的性质,研究者会使用不同级别的理论方法,如半经验方法、从头算方法以及密度泛函理论等。
### 2.3 分子模型的坐标系统
在分子建模中,描述分子中每个原子位置时,需要使用到坐标系统。这是分子建模中不可或缺的基础元素。
#### 2.3.1 坐标系统的基本概念
坐标系统可以是直角坐标系(Cartesian coordinates),也可以是极坐标系(polar coordinates)。在直角坐标系中,每个原子的位置用三个坐标值(x, y, z)来表示,而在极坐标系中,位置则是用距离和角度来描述。
在分子建模软件中,通常使用直角坐标系,因为它更直观地对应于物理空间。然而,在某些特定的计算,如表面分析或者在处理具有球对称性的分子系统时,极坐标系更为方便。
```mermaid
flowchart LR
A[坐标系统选择] -->|常规| B[直角坐标系]
A -->|特定计算| C[极坐标系]
B -->|优点| D[直观对应物理空间]
C -->|优点| E[方便处理球对称系统]
```
#### 2.3.2 直角坐标和极坐标的使用
在实际的分子建模中,直角坐标系使用更为广泛,因为它可以明确地描述每个原子在三维空间中的精确位置。直角坐标系的使用需要指定每个原子的x、y、z三个坐标值,这在计算机程序中通常表示为一个三元组。
例如,在一个简单的水分子(H2O)模型中,氧原子位于原点(0,0,0),两个氢原子分别位于x轴的正负方向,可以表示为(0,0,1)和(0,0,-1)。而极坐标系在描述球形分子或表面性质时更为方便,因为可以更简单地表示原子或分子片段在球面上的位置。
通过这一章节的学习,读者可以深入理解分子建模的科学基础,包括分子几何、化学键类型和坐标系统的选择。这些知识是建立有效分子模型的基础,对于进一步深入使用分子建模软件如Avogadro至关重要。下一章节将介绍Avogadro软件的基本操作技巧,包括界面布局、工具使用、分子的创建与编辑以及分子可视化等。
# 3. Avogadro基本操作技巧
在第二章中,我们了解了分子建模的基础理论,并深入探讨了分子几何、化学键以及分子模型的坐标系统。本章节将带您深入学习Avogadro软件的基本操作技巧,包括界面布局、工具使用、分子的创建与编辑以及分子可视化与渲染。通过本章节的介绍,读者将能够熟练操作Avogadro软件,为进一步的分子建模与分析打下坚实的基础。
## 3.1 界面布局与工具使用
### 3.1.1 Avogadro界面介绍
Avogadro软件界面布局简洁直观,主要分为菜单栏、工具栏、主视图区域以及状态栏几个部分。菜单栏提供了文件管理、编辑、视图、插件等多种功能。工具栏则提供常用功能的快捷操作,如新建文件、打开文件、保存、撤销等。主视图区域是用户进行分子建模和查看的主要

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
《Avogadro中文教程》专栏是一份全面的指南,旨在帮助读者掌握Avogadro软件的各个方面。从分子建模基础到高级应用,该专栏提供了深入的教程和技巧,让读者能够充分利用Avogadro的功能。
专栏涵盖了广泛的主题,包括个性化界面、工作效率提升、动力学模拟、数据分析、分子可视化、分子属性计算、量子化学集成、对称性与群论应用、药物设计、材料科学和生物学研究。通过一系列易于理解的教程和示例,该专栏旨在帮助读者充分利用Avogadro的强大功能,从而提高其在分子建模、计算化学和科学研究领域的效率和准确性。
专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
FANUC宏程序的自定义功能:扩展命令与创建个性化指令的技巧
# 摘要
本论文首先对FANUC宏程序的基础知识进行了概述,随后深入探讨了宏程序中扩展命令的原理,包括其与标准命令的区别、自定义扩展命令的开发流程和实例分析。接着,论文详细介绍了如何创建个性化的宏程序指令,包括设计理念、实现技术手段以及测试与优化方法。第四章讨论了宏程序的高级应用技巧,涉及错误处理、模块化与代码复用,以及与FANUC系统的集成。最后,论文探讨了宏程序的维护与管理问题,包括版本控制、文档化和知识管理,并对FANUC宏程序在先进企业的实践案例进行了分析,展望了技术的未来发展趋势。
# 关键字
FANUC宏程序;扩展命令;个性化指令;错误处理;模块化;代码复用;维护管理;技术趋势
【集成电路设计标准解析】:IEEE Standard 91-1984在IC设计中的作用与实践
# 摘要
本文系统性地解读了IEEE Standard 91-1984标准,并探讨了其在集成电路(IC)设计领域内的应用实践。首先,本文介绍了集成电路设计的基础知识和该标准产生的背景及其重要性。随后,文章详细分析了标准内容,包括设计流程、文档要求以及测试验证规定,并讨论了标准对提高设计可靠性和规范化的作用。在应用实践方面,本文探讨了标准化在设计流程、文档管理和测试验证中的实施,以及它如何应对现代IC设计中的挑战与机遇。文章通过案例研究展示了标准在不同IC项目中的应用情况,并分析了成功案例与挑战应对。最后,本文总结了标准在IC设计中的历史贡献和现实价值,并对未来集成电路设计标准的发展趋势进行了展
【中间件使用】:招行外汇数据爬取的稳定与高效解决方案
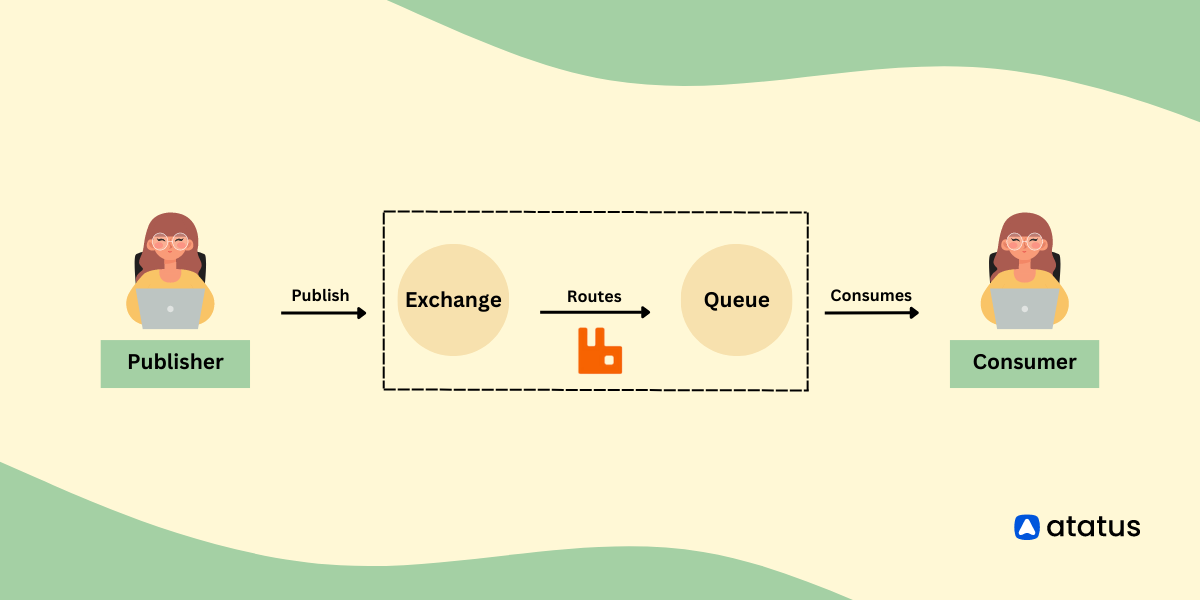
# 摘要
本文旨在探究外汇数据爬取技术及其在招商银行的实际应用。第一章简要介绍了中间件技术,为后续章节的数据爬取实践打下理论基础。第二章详细阐述了外汇数据爬取的基本原理和流程,同时分析了中间件在数据爬取过程中的关键作用及其优势。第三章通过招商银行外汇数据爬取实践,讨论了中间件的选择、配置以及爬虫稳定性与效率的优化方法。第四章探讨了分布式爬虫设计与数据存储处理的高级应用,
【带宽管理,轻松搞定】:DH-NVR816-128网络流量优化方案
# 摘要
本文对DH-NVR816-128网络流量优化进行了系统性的探讨。首先概述了网络流量的理论基础,涵盖了网络流量的定义、特性、波动模式以及网络带宽管理的基本原理和性能指标评估方法。随后,文章详细介绍了DH-NVR816-128设备的配置和优化实践,包括设备功能、流量优化设置及其在实际案例中的应用效果。文章第四章进一步探讨
easysite缓存策略:4招提升网站响应速度

# 摘要
网站响应速度对于用户体验和网站性能至关重要。本文探讨了缓存机制的基础理论及其在提升网站性能方面的作用,包括缓存的定义、缓存策略的原理、数据和应用缓存技术等。通过分析easysite的实际应用案例,文章详细阐述了缓存策略的实施步骤、效果评估以及监控方法。最后,本文还展望了缓存策略的未来发展趋势和面临的挑战,包括新兴缓存技术的应用以及云计算环境下缓存策略的创新,同时关注缓存策略实施过程中的安全性问
Impinj用户权限管理:打造强大多级权限系统的5个步骤
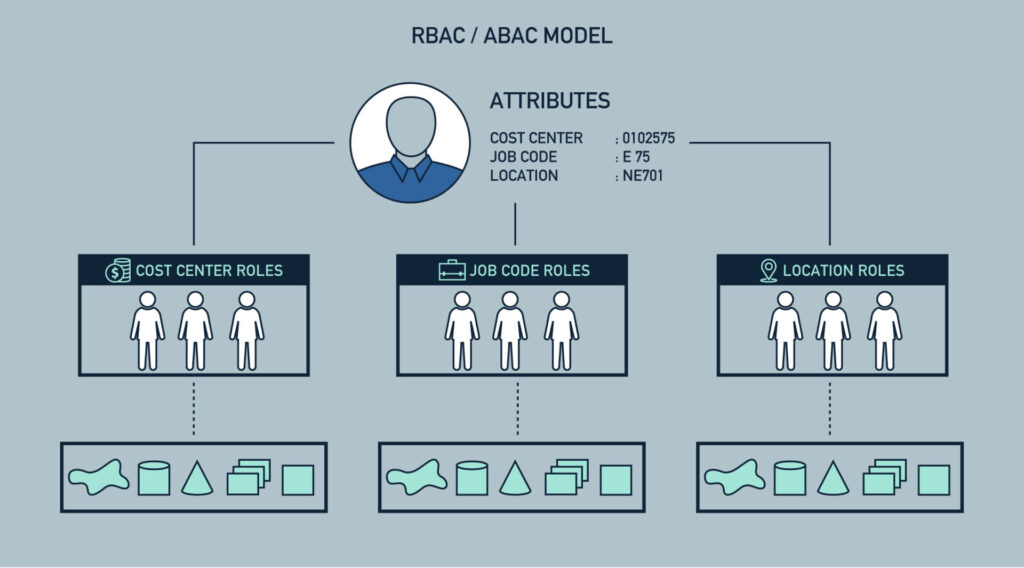
# 摘要
本文对Impinj权限管理系统进行了全面的概述与分析,强调了权限系统设计原则的重要性并详细介绍了Impinj权限模型的构建。通过深入探讨角色与权限的分配方法、权限继承机制以及多级权限系统的实现策略,本文为实现高效的权限控制提供了理论与实践相结合的方法。文章还涉及了权限管理在
北斗用户终端的设计考量:BD420007-2015协议的性能评估与设计要点
# 摘要
北斗用户终端作为北斗卫星导航系统的重要组成部分,其性能和设计对确保终端有效运行至关重要。本文首先概述了北斗用户终端的基本概念和特点,随后深入分析了BD420007-2015协议的理论基础,包括其结构、功能模块以及性能指标。在用户终端设计方面,文章详细探讨了硬件和软件架构设计要点,以及用户界面设计的重要性。此外,本文还对BD420007-2015协议进行了性能评估实践,搭建了测试环境,采用了基准测试和场景模拟等方法论,提出了基于评估结果的优化建议。最后,文章分析了北斗用户终端在不同场景下的应用,并展望了未来的技术创新趋势和市场发展策略。
# 关键字
北斗用户终端;BD420007-2
DS8178扫描枪图像处理秘籍:如何获得最清晰的扫描图像

# 摘要
本文全面介绍了图像处理的基础知识,聚焦DS8178扫描枪的硬件设置、优化与图像处理实践。文章首先概述了图像处理的基础和DS8178扫描枪的特性。其次,深入探讨了硬件设置、环境配置和校准方法,确保扫描枪的性能发挥。第三章详述了图像预处理与增强技术,包括噪声去除、对比度调整和色彩调整,以及图像质量评估方法。第四章结合实际应用案例,展示了如何优化扫描图像的分辨率和使用高级图像处理技术。最后,第五章介绍了
SW3518S芯片电源设计挑战:解决策略与行业最佳实践

# 摘要
本文综述了SW3518S芯片的电源设计理论基础和面临的挑战,提供了解决方案以及行业最佳实践。文章首先介绍了SW3518S芯片的电气特性和电源管理策略,然后着重分析了电源设计中的散热难题、能源转换效率和电磁兼容性问题。通过对实际案例的
批量安装一键搞定:PowerShell在Windows Server 2016网卡驱动安装中的应用

# 摘要
本文详细探讨了PowerShell在Windows Server环境中的应用,特别是在网卡驱动安装和管理方面的功能和优势。第一章概括了PowerShell的基本概念及其在Windows Server中的核心作用。第二章深入分析了网卡驱动安装的需求、挑战以及PowerShell自动
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )