【Chem3D案例研究】:用Chem3D解决实际化学问题的3个步骤
发布时间: 2025-01-06 17:16:04 阅读量: 15 订阅数: 10 

rip宣告网段选择版本

# 摘要
Chem3D软件是一款广泛应用于化学领域进行分子建模和模拟的工具。本文首先介绍了Chem3D的基本概念、安装流程以及其在化学模拟中的理论基础,包括化学模拟的重要性、分子建模和分子动力学模拟理论。随后,文章详细阐述了Chem3D在模拟操作实践中的应用,如分子建模与优化、模拟设置、执行及结果分析。通过实际案例分析,探讨了Chem3D在有机化学反应、材料科学和生物分子模拟中的具体应用。最后,本文探索了Chem3D的高级功能,并对其未来发展趋势和所面临的挑战进行了展望。
# 关键字
Chem3D;化学模拟;分子建模;分子动力学;模拟操作;高级功能
参考资源链接:[Chem3D分子结构演示:键长、键角与三维视图详解](https://wenku.csdn.net/doc/2ovafscw7v?spm=1055.2635.3001.10343)
# 1. Chem3D软件简介及安装
## 1.1 Chem3D软件概述
Chem3D是一款由PerkinElmer公司开发的化学信息学软件,广泛应用于化学研究与教育领域。该软件不仅提供了用户友好的界面,还集成了多种化学模拟工具,支持从基础的分子建模到高级的分子动力学模拟等复杂计算。
## 1.2 安装前的系统要求
在安装Chem3D之前,需要确保计算机满足以下最低系统要求:操作系统应为Windows 10或更高版本,处理器至少为Intel Core i5级别,内存至少8GB,硬盘空间需求则取决于安装的内容。如果要运行复杂的模拟,建议使用更强的硬件配置。
## 1.3 安装步骤详解
1. 从PerkinElmer官方网站下载Chem3D安装包。
2. 双击运行下载的安装文件,按照提示选择安装路径。
3. 完成安装后,启动软件,输入正版激活码激活。
以上步骤是安装Chem3D的基本流程,安装过程中确保网络连接稳定,以免下载中断导致安装失败。
# 2. 由于字数限制,这里仅提供第二章中两个子章节的详细内容。
## 2.1 化学模拟的重要性与目的
### 2.1.1 理论模拟与实验研究的关系
化学模拟是理论化学研究的一个重要分支,它通过计算模型来预测分子、化学反应和材料的性质。与传统的实验研究相比,化学模拟具有成本低、速度快、可重复性强等特点。理论上,模拟可以在任意给定条件下进行,而实验条件往往受到实验室环境、仪器精度以及操作人员的技能水平等多种因素的限制。
理论模拟与实验研究之间存在着互补关系。实验研究可以为模拟提供数据验证和参数校准,而模拟又可以指导实验的设计,帮助解释实验中的现象,甚至预测实验可能遇到的问题。例如,在药物研发领域,化学模拟可以预测分子间的相互作用,从而指导实验合成和生物活性测试的设计。
### 2.1.2 Chem3D在化学模拟中的应用范围
Chem3D作为一款强大的化学模拟软件,能够广泛应用于多个化学领域。在有机化学领域,Chem3D可以帮助化学家构建和优化分子结构,模拟反应过程,并预测反应路径和产物。在材料科学领域,通过分子模拟可以探索新材料的性质,优化材料的制备工艺,预测材料的稳定性和性能。
此外,生物化学和药物化学也是Chem3D应用的重要领域。例如,可以模拟生物大分子如蛋白质和核酸的结构,探究它们的功能与结构之间的关系,以及药物分子与靶点蛋白之间的相互作用。Chem3D软件的这些应用可以极大地提高科研效率,缩短新药研发周期,降低研发成本。
## 2.2 分子建模基础
### 2.2.1 分子几何结构的表示方法
分子的几何结构是指分子中各个原子之间的空间排列。分子几何结构的准确表示是分子模拟的基础。在Chem3D中,通常采用原子坐标来描述分子的几何结构。原子坐标可以是直角坐标系中的笛卡尔坐标,也可以是球坐标系中的极坐标。
为了便于理解和分析,分子的几何结构还可以用键长、键角和二面角来表示。键长是指原子之间的距离,键角是键连接的三个原子之间的夹角,二面角则是通过四个原子形成的平面之间的夹角。这些参数是评价分子构型和进行能量优化的重要依据。
### 2.2.2 分子力场与能量最小化
分子力场是分子模拟中描述原子核和电子之间相互作用的数学模型,它是分子模拟的基础。分子力场包含了多种类型的势能项,如键伸缩势、键角弯曲势、二面角势、范德华作用力和静电力等。通过这些势能项的组合,可以在计算机上计算出分子体系的能量。
分子模拟中的能量最小化是指通过调整原子坐标使分子体系的能量达到最小值的过程。在Chem3D中,能量最小化是一种常用的优化技术,它可以调整分子的几何结构,使得体系处于能量最低的稳定状态。在进行能量最小化时,通常采用迭代算法,如共轭梯度法、牛顿法或模拟退火法等。每一步迭代计算出的能量梯度用来指导下一步原子坐标的调整方向和幅度。
请注意,以上内容仅为第二章部分章节的详细内容,完整的章节和整篇文章内容将包含更多细节和深入分析,以满足给定的字数和格式要求。
# 3. Chem3D模拟操作实践
## 3.1 分子建模与优化
在化学模拟中,分子建模与优化是构建模型并使能量达到最小化的过程。这一阶段是进行任何化学模拟分析的基础。
### 3.1.1 创建分子结构的方法
Chem3D 提供了多种创建分子结构的方法,包括从头构建、从分子库中导入和基于已知结构修改。
**从头构建**
用户可以通过 Chem3D 的绘图工具栏手动绘制分子结构。选择“Build”菜单,即可找到“New Molecule”选项来启动绘图。用户可以逐个原子地添加,选择相应的原子类型(碳、氢、氧等)和键型(单键、双键、三键)进行构建。
```mermaid
graph TD
A[开始绘制] --> B[选择原子类型]
B --> C[选择键型]
C --> D[添加原子和键]
D --> E[构建完成]
```
**从分子库导入**
Chem3D 允许用户从内置的分子库中直接导入分子,该库包括了常见分子的结构,如水分子、氨分子等。
```plaintext
使用步骤:
1. 打开Chem3D
2. 在“File”菜单选择“Open”
3. 在打开的对话框中选择“
```

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
《模型的进一步信息》专栏深入探讨了 Chem3D 软件的广泛功能,为化学建模提供全面的指南。专栏涵盖了从数据可视化到动态模拟、量子化学计算和材料科学等各个方面。读者可以了解如何使用 Chem3D 设计和分析化学实验、自动化模拟流程、提高分子模型的精确度、预测分子行为、掌握量子化学计算的基础和高级应用,以及定制 Chem3D 界面以满足他们的特定需求。专栏还提供了解决实际化学问题的案例研究和提升模拟效率的专家技巧。无论您是化学建模的新手还是经验丰富的用户,本专栏都能为您提供宝贵的见解和实用的技巧,帮助您充分利用 Chem3D 的强大功能。
专栏目录

文章持续更新中,敬请期待~
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
【机器学习突破】:随机森林算法的深度解读及优化技巧

# 摘要
随机森林算法作为一种集成学习技术,在解决分类和回归任务中表现出色,尤其在数据挖掘、生物信息学和金融风险评估等领域应用广泛。本文首先概述了随机森林的基本概念及其理论基础,探讨了决策树的构建和剪枝策略,以及随机森林的工作原理和分类回归任务中的
射频系统中的LLCC68:信号完整性与干扰控制的秘技

# 摘要
本文系统介绍了LLCC68射频系统及其在信号完整性与干扰控制中的关键应用。首先概述了射频系统的基础知识和信号完整性的重要性,随后详细探讨了信号完整性分析工具和干扰控制的理论与实践。文
Keysight 34461A操作宝典:快速提升你的测量技能
# 摘要
Keysight 34461A多功能表是一款性能卓越的精密测量仪器,广泛应用于电子测试领域。本文首先概述了该仪器的基本特性和功能,接着介绍了测量的基础知识、工作原理、误差分析及提高数据精度的方法。第三章深入探讨了Keysight 34461A的各种测量功能,包括直流和交流电压电流测量以及电阻、电容和电感的高级测量。文章还具体阐述了如何操作实践,包括设备的连接、初始化、测量设置、参数调整及数据导出。最后,提供了一系列故障排除方法、维护指南以及高级应用技巧,确保用户能够高效利用仪器并处理常见问题。本论文旨在为电子测量技术提供全面的理论与实践指导,帮助工程师和技术人员更好地掌握和应用Key
CMG软件性能调优:专家告诉你如何提升系统效率
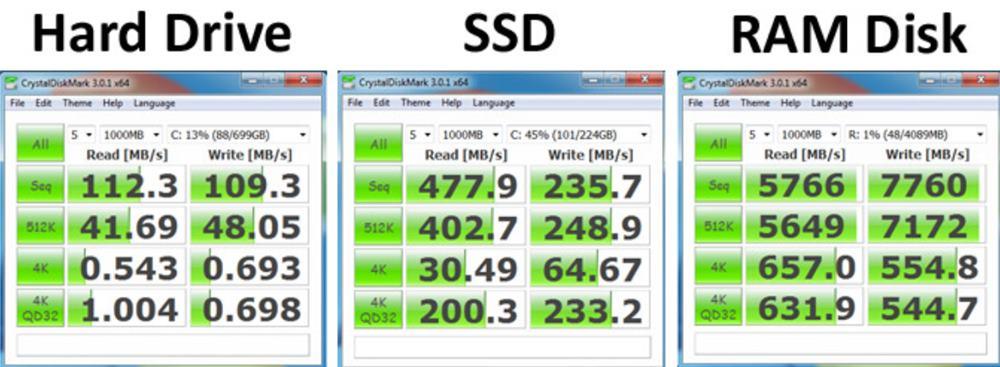
# 摘要
性能调优是确保软件应用高效运行的关键环节。本文首先介绍了性能调优的基础知识和CMG软件的基本概述,然后深入探讨了性能调优的核心理论,包括性能瓶颈识别、性能指标的确定以及CMG软件架构和性能指标的分析。在第三章中,本文详细论述了CMG软件监控和分析的方法,包括系统监控工具、日志分析以及CMG自带的性能分析工具的使用。第四章阐述了性能调优的实践策略,从调优前准备、
【报表性能提升攻略】:5种方法加速你的FastReport.NET报表加载与渲染
# 摘要
随着企业数据量的日益增长,报表的性能优化成为提升工作效率和用户体验的关键。本文首先强调了报表性能优化的重要性,并深入探讨了FastReport.NET报表引擎的核心原理、架构以及数据处理和渲染机制。接着,文章详细分析了报表加载性能提升的策略,
数据库系统原理:山东专升本,所有知识点一文搞定!
# 摘要
数据库系统作为信息管理的核心技术,涉及到数据的存储、处理和检索等关键操作。本文全面阐述了数据库系统的基础概念、核心组件,以及设计规范化与实践案例。深入讨论了数据库管理系统(DBMS)的三级模式架构,以及SQL语言在数据操作与查询中的应用。同时,探讨了数据库的规范化理论和设计方法论,包括需求分析、概念设计、逻辑设计与物理设计。此外,本文还涵盖了数据库系统的高级特性,如事务管理、并发控制、备份与恢
【编程新手必看】:微机原理课程设计指导,构建用户友好的打字计时器
# 摘要
微机原理课程设计旨在引导学生理解和掌握微机系统的基本结构与工作原理,尤其是在打字计时器的理论与实践应用中。本文首先概述了微机原理课程设计的重要性,继而详细阐述了打字计时器设计的理论基础,包括CPU和内存的基本概念、输入输出系统工作方式及用户界面需求。在设计与开发部分,重点介绍了系统架构、用户界面、硬件选择及连接、定时器模块设计。实现技术章节涉及编程技术、代码实现、调试与测试方法。实践操作章节则
案例深度剖析:如何利用SL651-2014规约解决水文监测中的实际问题

# 摘要
本文旨在详细介绍SL651-2014规约,阐述其理论基础、在水文监测系统中的应用实践以及高级应用和案例分析。文章首先对SL651-2014规约标准进行了详细解读,并结合水文监测的基础知识和数据采集传输过程,探讨了规约的核心内容和结构。其次,文章展示了规约在水文数据通信、监测设备配置以及数据
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
文章持续更新中,敬请期待~
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )