基因表达数据分析:从RNA-seq到功能注释
发布时间: 2024-01-16 22:35:29 阅读量: 107 订阅数: 43 
# 1. 引言
## 1.1 什么是基因表达数据分析
基因表达数据分析是对生物样本中各个基因的转录水平进行研究和解释的一项重要任务。通过分析基因表达数据,我们可以了解到不同组织或条件下基因的表达量变化情况,进而推断其在生物体内的功能和调控机制。基因表达数据分析在生物医学研究、植物育种、环境监测等领域都有广泛的应用。
## 1.2 RNA-seq技术简介
RNA-seq(RNA sequencing)是一种高通量测序技术,用于获取样本中所有转录的RNA序列。相比传统的基因表达分析方法,如Microarray,RNA-seq具有更高的灵敏度和更广泛的动态范围,能够检测到更多的基因表达变化,并发现新的基因型和转录本。
RNA-seq的基本流程包括:样本准备、RNA提取、RNA库构建、高通量测序和数据分析。数据分析是RNA-seq研究中的关键一步,主要包括数据预处理、差异基因分析、创新技术与工具应用、数据整合与挖掘等内容。
在本文中,我们将介绍基因表达数据分析的基本流程和常用方法。我们会详细讨论数据预处理、差异基因分析、创新技术与工具的应用以及数据整合与挖掘的方法。最后,我们将总结当前领域的一些挑战和未来发展的方向。
# 2. 数据预处理
在进行基因表达数据分析之前,首先需要对原始数据进行预处理。数据预处理包括质量控制和过滤、序列比对与定量以及基本统计学分析。这些步骤旨在去除噪音数据、规范化数据格式,并为后续的差异基因分析做准备。
### 2.1 质量控制和过滤
质量控制和过滤是数据预处理的重要步骤,其目的是确保实验数据的准确性和可靠性。常用的质量控制工具包括FastQC、Trim Galore等。以下是一个使用Trim Galore进行质量控制和过滤的示例代码:
```python
import subprocess
def quality_control(input_file, output_dir):
subprocess.run(['trim_galore', '--quality', '20', '--output_dir', output_dir, input_file])
# 调用质量控制函数
input_file = 'raw_data.fastq'
output_dir = 'qc_output'
quality_control(input_file, output_dir)
```
在上述代码中,我们使用了trim_galore工具进行质量控制和过滤,设定了质量阈值为20,并将输出目录设置为qc_output。
### 2.2 序列比对与定量
在数据预处理阶段,我们需要将数据进行序列比对和基因定量。常用的比对工具包括Bowtie、TopHat、HISAT等,而基因定量工具则包括HTSeq、featureCounts等。以下是一个使用Bowtie进行序列比对的示例代码:
```python
import subprocess
def sequence_alignment(input_file, output_file):
bowtie_index = 'genome_index'
subprocess.run(['bowtie', bowtie_index, input_file, output_file])
# 调用序列比对函数
input_file = 'clean_data.fastq'
output_file = 'alignment.sam'
sequence_alignment(input_file, output_file)
```
在上述代码中,我们使用了Bowtie工具进行序列比对,指定了参考基因组的索引文件为genome_index,并将比对结果保存为alignment.sam文件。
### 2.3 基本统计学分析
在数据预处理之后,我们可以进行一些基本的统计学分析来了解数据的特征。常见的统计学分析包括计算测序深度、基因表达量的分布情况等。以下是一个计算测序深度的示例代码:
```python
import pysam
def calculate_read_depth(input_file):
samfile = pysam.AlignmentFile(input_file, 'r')
total_reads = samfile.count()
average_depth = total_reads / samfile.header['SQ'][0]['LN']
samfile.close()
return average_depth
# 调用计算测序深度函数
input_file = 'alignment.sam'
read_depth = calculate_read_depth(input_file)
print("Average read depth: ", read_depth)
```
在上述代码中,我们使用了pysam库来读取比对文件,并计算了每个碱基的平均测序深度。最后,我们将平均测序深度输出到屏幕上。
数据预处理阶段的质量控制和过滤、序列比对与定量以及基本统计学分析的步骤能够确保后续的差异基因分析的准确性和可靠性。
# 3. 差异基因分析
基因表达数据分析的一个重要方面是对不同条件下基因表达水平的差异进行分析,以揭示基因在特定生物学过程或疾病状态中的重要作用。在这一章节中,我们将介绍差异基因分析的相关内容,包括差异表达基因的检测和筛选、功能富集分析以及可视化与结果解释。
#### 3.1 差异表达基因的检测和筛选
差异表达基因分析是基因表达数据分析的核心环节之一,通过比较不同样本条件下的基因表达水平,识别在不同条件间表达水平存在显著差异的基因。常用的方法包括DESeq2、edgeR等,在这里我们以Python中的DESeq2为例进行示范。
```python
# 导入DESeq2库
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
from scipy import stats
from statsmodels.stats import multitest
im
```

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
本专栏《生物数据分析与信息处理技术:生物信息学与基因组学应用》旨在通过一系列文章深入介绍生物信息学与基因组学的相关概念和应用技术。专栏包括了DNA序列分析入门、BLAST算法的应用、基因表达数据分析、重复序列分析、基因预测技术比较、蛋白质序列分析、生物数据存储与管理、基于NGS的变异检测技术、功能基因组学的元件识别、ChIP-seq技术与染色质免疫沉淀数据分析、元转录组学在微生物研究中的应用等多个主题。此外,专栏还着眼于代谢组学数据处理、药物基因组学以及DNA条形码研究等前沿领域。通过本专栏,读者将能够全面了解生物数据分析与信息处理技术在生物学研究中的重要性和应用价值,为相关领域的学习和实践提供全面的指导和启发。
最低0.47元/天 解锁专栏
买1年送1年
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
【MATLAB在Pixhawk定位系统中的应用】:从GPS数据到精确定位的高级分析

# 1. Pixhawk定位系统概览
Pixhawk作为一款广泛应用于无人机及无人车辆的开源飞控系统,它在提供稳定飞行控制的同时,也支持一系列高精度的定位服务。本章节首先简要介绍Pixhawk的基本架构和功能,然后着重讲解其定位系统的组成,包括GPS模块、惯性测量单元(IMU)、磁力计、以及_barometer_等传感器如何协同工作,实现对飞行器位置的精确测量。
我们还将概述定位技术的发展历程,包括
面向对象编程:继承机制的终极解读,如何高效运用继承提升代码质量

# 1. 面向对象编程中的继承机制
面向对象编程(OOP)是一种编程范式,它使用“对象”来设计软件。这些对象可以包含数据,以字段(通常称为属性或变量)的形式表示,以及代码,以方法的形式表示。继承机制是OOP的核心概念之一,它允许新创建的对象继承现有对象的特性。
## 1.1 继承的概念
继承是面向对象编程中的一个机制,允许一个类(子类)继承另一个类(父类)的属性和方法。通过继承
消息队列在SSM论坛的应用:深度实践与案例分析

# 1. 消息队列技术概述
消息队列技术是现代软件架构中广泛使用的组件,它允许应用程序的不同部分以异步方式通信,从而提高系统的可扩展性和弹性。本章节将对消息队列的基本概念进行介绍,并探讨其核心工作原理。此外,我们会概述消息队列的不同类型和它们的主要特性,以及它们在不同业务场景中的应用。最后,将简要提及消息队列
MATLAB时域分析:动态系统建模与分析,从基础到高级的完全指南
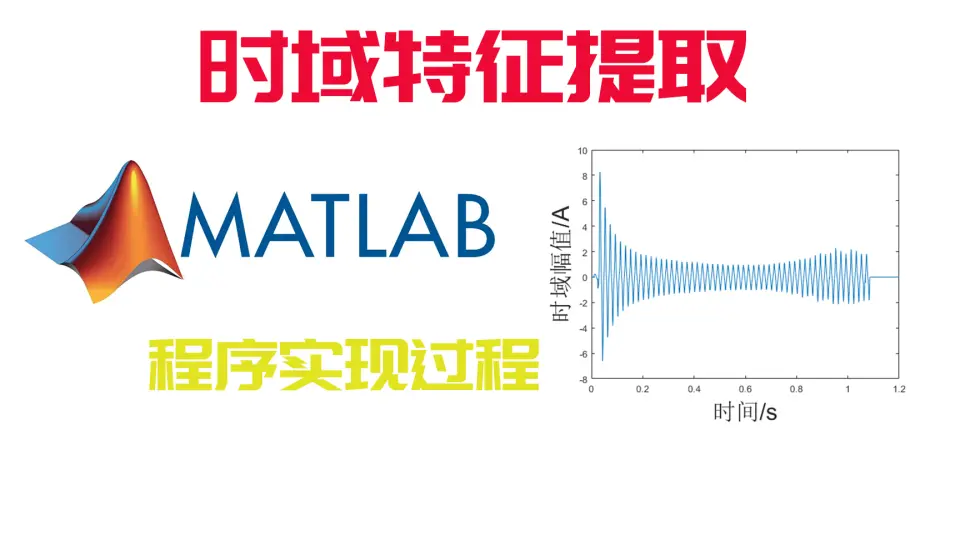
# 1. MATLAB时域分析概述
MATLAB作为一种强大的数值计算与仿真软件,在工程和科学领域得到了广泛的应用。特别是对于时域分析,MATLAB提供的丰富工具和函数库极大地简化了动态系统的建模、分析和优化过程。在开始深入探索MATLAB在时域分析中的应用之前,本章将为读者提供一个基础概述,包括时域分析的定义、重要性以及MATLAB在其中扮演的角色。
时域
【大数据处理利器】:MySQL分区表使用技巧与实践
# 1. MySQL分区表概述与优势
## 1.1 MySQL分区表简介
MySQL分区表是一种优化存储和管理大型数据集的技术,它允许将表的不同行存储在不同的物理分区中。这不仅可以提高查询性能,还能更有效地管理数据和提升数据库维护的便捷性。
## 1.2 分区表的主要优势
分区表的优势主要体现在以下几个方面:
- **查询性能提升**:通过分区,可以减少查询时需要扫描的数据量
故障恢复计划:机械运动的最佳实践制定与执行

# 1. 故障恢复计划概述
故障恢复计划是确保企业或组织在面临系统故障、灾难或其他意外事件时能够迅速恢复业务运作的重要组成部分。本章将介绍故障恢复计划的基本概念、目标以及其在现代IT管理中的重要性。我们将讨论如何通过合理的风险评估与管理,选择合适的恢复策略,并形成文档化的流程以达到标准化。
## 1.1 故障恢复计划的目的
故障恢复计划的主要目的是最小化突发事件对业务的
【深度学习在卫星数据对比中的应用】:HY-2与Jason-2数据处理的未来展望

# 1. 深度学习与卫星数据对比概述
## 深度学习技术的兴起
随着人工智能领域的快速发展,深度学习技术以其强大的特征学习能力,在各个领域中展现出了革命性的应用前景。在卫星数据处理领域,深度学习不仅可以自动
Python讯飞星火LLM数据增强术:轻松提升数据质量的3大法宝
# 1. 数据增强技术概述
数据增强技术是机器学习和深度学习领域的一个重要分支,它通过创造新的训练样本或改变现有样本的方式来提升模型的泛化能力和鲁棒性。数据增强不仅可以解决数据量不足的问题,还能通过对数据施加各种变化,增强模型对变化的适应性,最终提高模型在现实世界中的表现。在接下来的章节中,我们将深入探讨数据增强的基础理论、技术分类、工具应用以及高级应用,最后展望数据增强技术的
Python调试技术速成课:快速定位问题的终极技巧
# 1. Python调试技术概述
## 简介
Python作为一门广受欢迎的高级编程语言,拥有众多的开发工具和库来支持开发者快速构建应用程序。然而,在任何复杂的开发过程中,代码的调试是不可避免的一部分,它是保证软件质量、提升开发效率的关键步骤。
## 调试技术的重要性
代码质量是软件开发的基石,而有效的调试技术能够帮助开发者发现和修复代码中的错误与缺陷。调试不仅仅是找出问题所在,更关键的是通过调试过程来
拷贝构造函数的陷阱:防止错误的浅拷贝
# 1. 拷贝构造函数概念解析
在C++编程中,拷贝构造函数是一种特殊的构造函数,用于创建一个新对象作为现有对象的副本。它以相同类类型的单一引用参数为参数,通常用于函数参数传递和返回值场景。拷贝构造函数的基本定义形式如下:
```cpp
class ClassName {
public:
ClassName(const ClassName& other); // 拷贝构造函数
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送1年
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )