基因表达数据聚类与分类方法
发布时间: 2024-02-03 20:43:27 阅读量: 85 订阅数: 23 

基因表达数据分类的混合特征选择算法
# 1. 引言
## 1.1 基因表达数据的重要性
基因表达数据是指记录了基因在生物体中的表达水平的数据,通过测量和记录基因在不同组织、不同时间点或不同环境条件下的表达水平,可以了解基因在生物体中的功能和调控机制。基因表达数据的获取方式主要有基因芯片、RNA测序和蛋白质质谱等多种方法,其中RNA测序是目前最常用的方法之一。
基因表达数据的重要性在于它可以帮助科研人员理解生物体内基因的功能和调控机制。通过对基因表达数据的分析,可以发现某些基因在特定组织或特定环境条件下的表达水平的变化,推断该基因可能在该组织或环境下具有特定的功能或参与特定的调控过程。此外,基因表达数据也可以用于研究疾病的发生机制和治疗方法的探索,通过比较疾病患者和健康人群的基因表达差异,可以找到与疾病相关的基因和通路,为疾病的诊断和治疗提供依据。
## 1.2 聚类与分类在基因表达数据中的应用价值
聚类和分类是基因表达数据分析中常用的数据挖掘方法,它们可以将基因表达数据中相似的样本或基因分组在一起,从而揭示出不同样本之间的关系和特点。聚类和分类在基因表达数据中的应用价值主要体现在以下几个方面。
首先,聚类和分类可以帮助科研人员对基因表达数据进行初步的分析和理解。通过聚类分析,可以将相似的样本或基因聚集在一起,帮助科研人员发现潜在的样本分类或基因簇。通过分类分析,可以将样本或基因按照一定标准进行分类,从而帮助科研人员理清数据的特点和结构。
其次,聚类和分类可以帮助科研人员发现基因表达数据中的模式和规律。通过聚类分析,可以发现不同样本之间的相似性和差异性,揭示出基因表达数据中可能存在的模式和规律。通过分类分析,可以发现不同基因之间的相关性和相互作用,揭示基因表达数据中的调控网络和通路。
最后,聚类和分类可以帮助科研人员进行基因表达数据的预测和分类。通过聚类分析,可以将新样本归入已有的样本簇中,从而预测新样本的特征和性质。通过分类分析,可以将新样本分类到已有的类别中,从而对新样本进行分类和识别。
综上所述,聚类和分类在基因表达数据分析中具有重要的应用价值,可以帮助科研人员对基因表达数据进行初步的分析和理解,发现数据中的模式和规律,并进行预测和分类。在接下来的文章中,我们将介绍基因表达数据的聚类方法和分类方法,并通过实例研究展示它们的应用效果。
# 2. 基因表达数据的聚类方法
基因表达数据的聚类方法是将基因表达矩阵中的基因或样本进行分组,使得同一组内的基因或样本具有相似的表达模式。聚类方法有助于揭示基因表达数据中的潜在模式和结构,为后续的生物信息学分析提供重要线索。
### 2.1 层次聚类
层次聚类是一种基于相似度或距离的聚类方法,通过逐渐合并或划分样本或基因来构建聚类树。该方法适用于小样本量和较高维度的数据,能够直观地展现聚类结果。常见的层次聚类算法包括基于距离的最小值(single-linkage)、最大值(complete-linkage)和平均值(average-linkage)等。
```python
# Python代码示例:使用scipy库进行层次聚类
from scipy.cluster.hierarchy import linkage, dendrogram
import matplotlib.pyplot as plt
import numpy as np
# 生成随机基因表达数据
data = np.random.rand(10, 5)
# 计算距离矩阵
linkage_matrix = linkage(data, method='complete')
# 绘制聚类树状图
dendrogram(linkage_matrix)
plt.show()
```
### 2.2 k均值聚类
k均值聚类是一种基于距离的分区聚类方法,将样本划分为k个簇,使得同一簇内样本之间的距离最小化。该方法适用于大型数据集和均匀分布的簇结构。然而,k均值聚类对初始聚类中心的选择敏感,且需要事先确定k的取值。
```java
// Java代码示例:使用weka库进行k均值聚类
import weka.clusterers.SimpleKMeans;
import weka.core.Instances;
import weka.core.converters.ConverterUtils.DataSource;
// 读取基因表达数据
DataSource source = new DataSource("gene_expression.arff");
Instances data = source.getDataSet();
// 初始化k均值聚类器
SimpleKMeans kmeans = new SimpleKMeans();
kmeans.setNumClusters(3);
kmeans.buildClusterer(data);
```
### 2.3 密度聚类
密度聚类是一种基于样本密度的聚类方法,通过识别样本密度大于给定阈值的核心对象,并将与核心对象密度可达的样本归为同一簇。相比于传统的基于距离的聚类方法,密度聚类能够适应不规则形状的簇结构。
```go
// Go代码示例:使用go-cluster库进行DBSCAN密度聚类
import "github.com/mpraski/clusters"
import "github.com/gonum/matrix/mat64"
// 生成基因表达数据
data := mat64.NewDense(10, 5, nil)
// 初始化DBSCAN聚类器
dbscan := clusters.NewDBSCAN(0.5, 2)
clusters := dbscan.Clusterize(data)
```
### 2.4 谱聚类
谱聚类是一种基于图论的聚类方法,通过样本之间的相似度构建相似度矩阵,进而利用特征值分解等方法对相似度矩阵进行降维和聚类。该方法能够有效处理非凸形状的簇结构,并且不需要预先指定簇的个数。
```javascript
// JavaScript代码示例:使用ml-kmeans库进行谱聚类
const ml = require('ml-kmeans');
const data = [[1, 2, 3], [4, 5, 6], [7, 8, 9]];
// 调用谱聚类算法
const result = ml.kmeans(data, 2);
console.log(result.clusters);
```
### 2.5 优缺点比较与选择
不同的聚类方法具有各自的优缺点,例如层次聚类对异常值不敏感但计算复杂度较高,k均值聚类对初始值敏感但计算速度快。在实际应用中,需要根据数据特点和聚类目的进行选择,并结合交叉验证等方法进行优化调参。
# 3. 基因表达数据的分类方法
在基因表达数据中,分类是一项重要的任务,它可以将样本分为不同的类别,从而揭示不同基因在不同生物学条件下的表达模式。下面介绍几种常见的基因表达数据分类方法。
#### 3.1 逻辑回归
逻辑回归是一种广泛应用于分类问题的统计方法。在基因表达数据分类中,逻辑回归可以用于预测样本所属的类别。逻辑回归的主要思想是通过

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
本专栏以"生物数据分析与信息处理方法基础与应用"为主题,涵盖了生物信息学领域中的多个重要主题。从生物数据的预处理技术到基因组测序、转录组学、蛋白质组学、生物网络分析、基因表达数据聚类分类、生物序列比对与注释、生物图像分析与处理、单细胞测序、DNA甲基化数据分析、功能富集分析、介观基因组学、深度学习应用、代谢组学数据分析、蛋白质结构预测、基因调控网络建模等方面进行了深入解析。每篇文章均以介绍最新的理论与方法为主,并结合真实案例进行应用展示。该专栏旨在帮助读者全面了解生物数据分析与信息处理领域的最新进展,为生物学、医学以及生命科学领域的从业者提供专业的学习与参考。
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
扇形菜单高级应用

# 摘要
扇形菜单作为一种创新的用户界面设计方式,近年来在多个应用领域中显示出其独特优势。本文概述了扇形菜单设计的基本概念和理论基础,深入探讨了其用户交互设计原则和布局算法,并介绍了其在移动端、Web应用和数据可视化中的应用案例
C++ Builder高级特性揭秘:探索模板、STL与泛型编程

# 摘要
本文系统性地介绍了C++ Builder的开发环境设置、模板编程、标准模板库(STL)以及泛型编程的实践与技巧。首先,文章提供了C++ Builder的简介和开发环境的配置指导。接着,深入探讨了C++模板编程的基础知识和高级特性,包括模板的特化、非类型模板参数以及模板
【深入PID调节器】:掌握自动控制原理,实现系统性能最大化

# 摘要
PID调节器是一种广泛应用于工业控制系统中的反馈控制器,它通过比例(P)、积分(I)和微分(D)三种控制作用的组合来调节系统的输出,以实现对被控对象的精确控制。本文详细阐述了PID调节器的概念、组成以及工作原理,并深入探讨了PID参数调整的多种方法和技巧。通过应用实例分析,本文展示了PID调节器在工业过程控制中的实际应用,并讨
【Delphi进阶高手】:动态更新百分比进度条的5个最佳实践

# 摘要
本文针对动态更新进度条在软件开发中的应用进行了深入研究。首先,概述了进度条的基础知识,然后详细分析了在Delphi环境下进度条组件的实现原理、动态更新机制以及多线程同步技术。进一步,文章探讨了数据处理、用户界面响应性优化和状态视觉呈现的实践技巧,并提出了进度
【TongWeb7架构深度剖析】:架构原理与组件功能全面详解
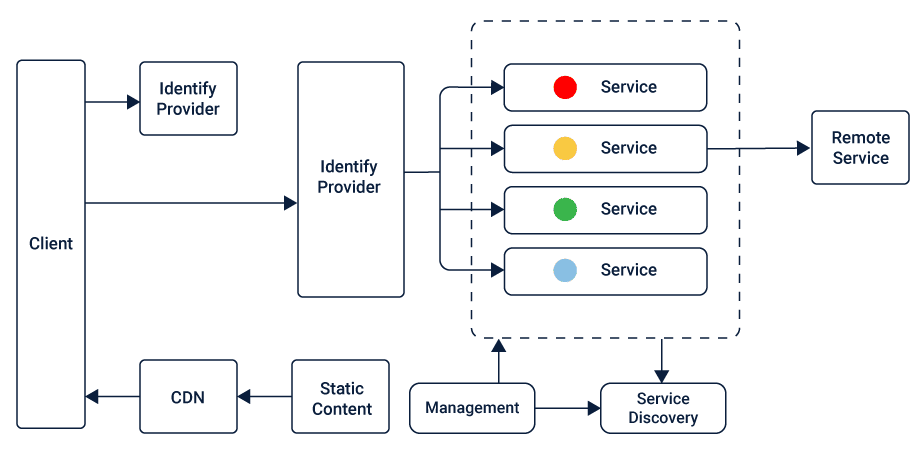
# 摘要
TongWeb7作为一个复杂的网络应用服务器,其架构设计、核心组件解析、性能优化、安全性机制以及扩展性讨论是本文的主要内容。本文首先对TongWeb7的架构进行了概述,然后详细分析了其核心中间件组件的功能与特点,接着探讨了如何优化性能监控与分析、负载均衡、缓存策略等方面,以及安全性机制中的认证授权、数据加密和安全策略实施。最后,本文展望
【S参数秘籍解锁】:掌握驻波比与S参数的终极关系
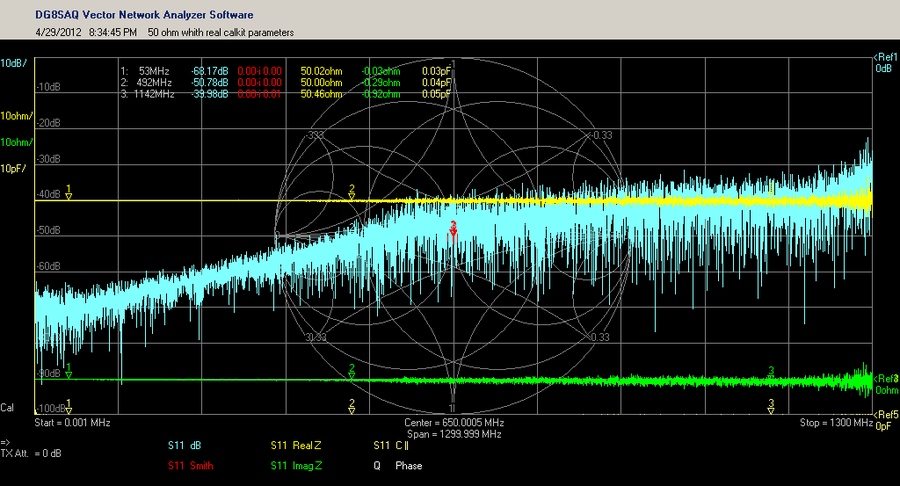
# 摘要
本论文详细阐述了驻波比与S参数的基础理论及其在微波网络中的应用,深入解析了S参数的物理意义、特性、计算方法以及在电路设计中的实践应用。通过分析S参数矩阵的构建原理、测量技术及仿真验证,探讨了S参数在放大器、滤波器设计及阻抗匹配中的重要性。同时,本文还介绍了驻波比的测量、优化策略及其与S参数的互动关系。最后,论文探讨了S参数分析工具的使用、高级分析技巧,并展望
【嵌入式系统功耗优化】:JESD209-5B的终极应用技巧
# 摘要
本文首先概述了嵌入式系统功耗优化的基本情况,随后深入解析了JESD209-5B标准,重点探讨了该标准的框架、核心规范、低功耗技术及实现细节。接着,本文奠定了功耗优化的理论基础,包括功耗的来源、分类、测量技术以及系统级功耗优化理论。进一步,本文通过实践案例深入分析了针对JESD209-5B标准的硬件和软件优化实践,以及不同应用场景下的功耗优化分析。最后,展望了未来嵌入式系统功耗优化的趋势,包括新兴技术的应用、JESD209-5B标准的发展以及绿色计算与可持续发展的结合,探讨了这些因素如何对未来的功耗优化技术产生影响。
# 关键字
嵌入式系统;功耗优化;JESD209-5B标准;低功耗
ODU flex接口的全面解析:如何在现代网络中最大化其潜力
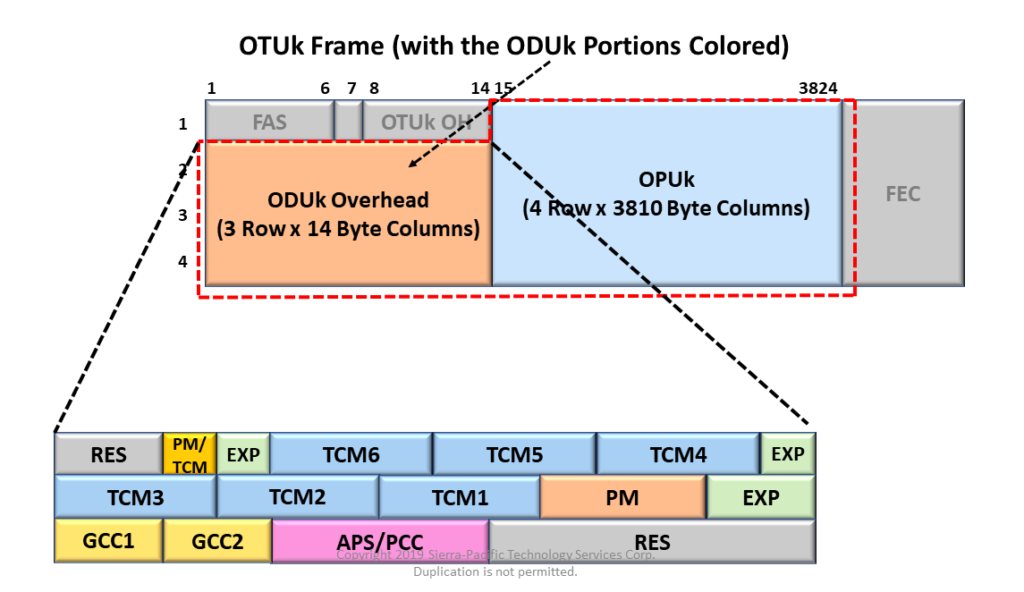
# 摘要
ODU flex接口作为一种高度灵活且可扩展的光传输技术,已经成为现代网络架构优化和电信网络升级的重要组成部分。本文首先概述了ODU flex接口的基本概念和物理层特征,紧接着深入分析了其协议栈和同步机制,揭示了其在数据中心、电信网络、广域网及光纤网络中的应用优势和性能特点。文章进一步
如何最大化先锋SC-LX59的潜力
# 摘要
先锋SC-LX59作为一款高端家庭影院接收器,其在音视频性能、用户体验、网络功能和扩展性方面均展现出巨大的潜力。本文首先概述了SC-LX59的基本特点和市场潜力,随后深入探讨了其设置与配置的最佳实践,包括用户界面的个性化和音画效果的调整,连接选项与设备兼容性,以及系统性能的调校。第三章着重于先锋SC-LX59在家庭影院中的应用,特别强调了音视频极致体验、智能家居集成和流媒体服务的充分利用。在高
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )