量子过渡态计算:原理揭示与实际操作指南
发布时间: 2025-01-03 10:37:44 阅读量: 13 订阅数: 20 

# 摘要
量子过渡态计算是理解化学反应和材料科学中动态过程的关键,它在预测反应速率和产物分布方面具有重要意义。本文首先介绍了量子过渡态计算的基础和理论框架,强调了量子力学原理和过渡态理论在计算中的应用。接着,重点讨论了目前市场上主流的量子计算软件工具,包括它们的功能和操作方法,并展示了如何运用这些工具进行实验设计、模拟和结果分析。文章还探索了量子过渡态计算的前沿方向,如新兴的量子计算技术、过渡态理论的扩展以及量子计算与机器学习的结合。最后,通过实际案例分析了量子过渡态计算在工业和科研中的应用,并对未来发展趋势进行了展望。
# 关键字
量子过渡态计算;量子力学;能量势垒;过渡态理论;量子计算软件;机器学习
参考资源链接:[详解CINEB方法下的VASP过渡态计算步骤与VTST CI-NEB应用](https://wenku.csdn.net/doc/740w943acw?spm=1055.2635.3001.10343)
# 1. 量子过渡态计算基础
## 1.1 过渡态计算的意义
过渡态计算在化学反应动力学中扮演着至关重要的角色。它不仅帮助科学家理解反应的机制,而且对于预测反应速率以及优化化学过程至关重要。传统化学研究依赖于实验数据,而量子过渡态计算提供了一种计算上的途径,能够在分子层面上对反应过程进行模拟和分析。
## 1.2 量子过渡态计算的基本概念
量子过渡态计算是应用量子力学原理对化学反应中的过渡态进行研究的一种方法。过渡态可以被理解为反应物转化为产物的瞬间,此时系统处于能量的最大点。通过对过渡态的理解,我们可以更好地掌握反应过程中的电子行为和原子的位置变化。
## 1.3 过渡态计算的研究工具
为了进行量子过渡态计算,研究者们使用各种计算化学软件和工具。这些工具通过解决薛定谔方程来预测分子体系的行为。它们包括从简单的半经验方法到复杂的密度泛函理论(DFT)和从头算方法(ab initio)。使用这些工具,研究者们可以计算电子结构、能量变化,以及反应坐标上的势能面。
# 2. 量子过渡态理论框架
## 2.1 量子力学的数学基础
### 2.1.1 线性代数在量子力学中的应用
在量子力学领域,线性代数构成了数学语言的核心。量子态可由向量表示,在希尔伯特空间中,这些向量遵循特定的规则与操作。理解线性代数在量子力学中的应用,能够让我们更好地理解和操作量子态。
希尔伯特空间提供了一种描述量子系统所有可能状态的数学框架。量子态作为这个空间中的向量,需要遵循内积运算规则,其结果是复数。状态的内积代表了量子系统不同状态的重叠程度,而这正是量子力学中态叠加原理的基础。
量子力学的线性特性意味着,如果一个量子系统处于状态 |ψ⟩,那么该系统也处于任何与 |ψ⟩ 相关的线性组合的状态下,如 a|ψ⟩+b|φ⟩,其中 a 和 b 是复数系数。线性代数为量子系统的这种组合提供了明确的数学描述。
量子力学的可观测量,如位置、动量或能量,由厄米算符(Hermitian operators)表示。厄米算符在量子力学的数学模型中非常重要,因为它们具有实数特征值,对应于观测到的物理量。通过对厄米算符进行对角化,可以找到系统的本征态和本征值,这些是计算能量级等物理量的基础。
### 2.1.2 波函数与态叠加原理
波函数是量子力学中描述量子态的数学函数,通常用希腊字母 Ψ 表示。波函数包含了关于粒子位置、动量等信息的概率分布。通过波函数的绝对值的平方,我们能计算粒子出现在特定位置的概率密度。波函数的平方 |Ψ(x)|^2 dx 给出粒子在位置 x 到 x+dx 之间出现的概率。
态叠加原理说明了量子系统可以处于多个状态的“叠加”中。在波函数的语言中,这意味着一个量子系统可以被描述为多个波函数的线性组合,即 Ψ = Σc_iψ_i,其中 c_i 是复数系数,ψ_i 表示不同的量子态。当我们对这个系统进行测量时,它会“坍缩”到其中一个本征态,测量的结果对应于该本征态的概率幅的平方的绝对值。
为了深入理解波函数,我们必须要知道薛定谔方程。这个方程描述了量子态如何随时间演化。通过求解薛定谔方程,我们可以得到波函数随时间变化的具体形式,从而预测量子系统的行为。
## 2.2 过渡态理论概述
### 2.2.1 能量势垒与反应速率
过渡态理论的核心思想在于化学反应涉及从反应物到产物的能量势垒,而这个势垒是由过渡态(也称为活化复合物)所界定的。在这个过渡态中,反应物分子转化成产物分子所必须达到的高能量状态。这一理论对理解反应速率以及反应动力学提供了基础。
能量势垒的高低直接影响化学反应的速率。高势垒意味着需要更多的能量才能实现反应,因此反应速率较低。而低势垒意味着较小的能量输入就能驱动反应进行,从而具有较高的反应速率。因此,势垒的高度是决定反应速率的关键因素之一。
为了计算反应速率,过渡态理论引入了活化能的概念,这是指反应物分子变为活化复合物所需克服的能量。根据阿伦尼乌斯方程,反应速率常数与温度和活化能有直接关系。因此,活化复合物的确切性质对反应动力学有着决定性的影响。
### 2.2.2 过渡态与反应坐标
在反应过程中,系统经过一个能量极值点,这个点被称为过渡态。反应坐标是描述反应物向产物转化过程的一个概念性参数。它提供了一个方式来追踪反应进程,量化了反应物如何转化为产物。
一个完整的反应坐标图包括了从反应物到产物的能量变化,以及过渡态所处的特定位置。这种能量变化通常在所谓的“势能面”上表示,可以想象成一座山的轮廓,山峰代表过渡态,而山的两侧则分别是反应物和产物的势能。
了解反应坐标不仅可以预测反应路径和机制,还能揭示反应是否可逆,以及任何可能的中间产物。过渡态的识别是化学反应速率和机制研究的关键步骤,它让我们能够更精确地理解化学反应的动力学行为。
## 2.3 过渡态计算的方法论
### 2.3.1 传统过渡态计算方法
传统计算化学中,过渡态寻找基于势能面的扫描以及优化技术。这些方法包括分子动力学模拟、Monte Carlo模拟、以及基于线性变分方法的从头算和半经验计算。
分子动力学模拟通过解决牛顿运动方程来跟踪原子和分子随时间的运动,可以提供反应过程的直观图像。Monte Carlo方法利用随机抽样来计算系统的宏观性质,特别适用于处理大规模复杂系统。
线性变分方法是一种量子化学的计算方法,主要依赖于对波函数的假设,来计算体系的能量和波函数。从头算方法完全基于量子力学的第一原理,不依赖于实验参数,而半经验方法则结合了实验数据和理论计算,以减小计算的复杂性。
### 2.3.2 现代量子计算方法对比
随着计算能力的增强和理论的发展,量子过渡态计算正逐步采用现代方法,比如密度泛函理论(DFT),量子蒙特卡洛(QMC)和路径积分方法等。这些方法提供了更加精确和高效地预测化学反应的方式。
DFT是一种广泛应用于计算材料科学和化学领域的量子化学方法。它基于电子密度而非波函数,极大地简化了计算过程,同时仍能提供足够的精度。DFT的核心优势在于其对固体材料和大型分子系统的计算能力。
量子蒙特卡洛方法通过随机模拟来解决多体量子问题,尤其在处理电子关联时表现突出。它不依赖于传统的波函数展开,而是使用随机抽样技术来得到系统波函数和能量的统计描述。
路径积分方法是基于量子路径的概念,它提供了一种不同的视角来考虑量子行为。路径积分方法尤其适合于描述量子涨落和非局域性,因此在处理量子过渡态时具有特别的优势。
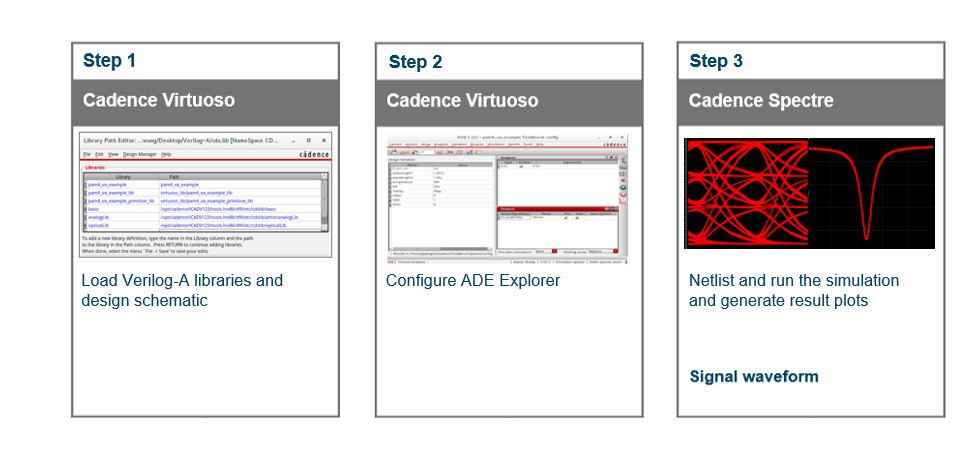
# 3. 量子过渡态计算工具和软件
## 3.1 量子计算软件综述

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
《过渡态计算终极指南》专栏深入探讨了过渡态计算这一强大工具,揭示了它在化学反应速率预测、材料研发、催化剂设计和生物分子动力学等领域的应用。从经典力学到量子化学,从理论原理到实践策略,专栏提供了全面的指南,帮助读者掌握过渡态模拟的精髓。此外,专栏还探讨了误差分析、高效并行算法、熵与自由能评估、速度和准确性优化等关键主题,为读者提供了全方位的知识和技能,以利用过渡态计算解锁化学反应的奥秘,推动科学和工业领域的创新。
专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
【高可用性与备份】:VCS备份路径方案确保数据安全的关键步骤

# 摘要
本文深入探讨了高可用性基础和备份策略的重要性、设计与实现,以及VCS基础和高可用性集群架构的关键组成。文章首先强调了备份在保障数据安全和系统稳定运行中的基础作用,随后详细介绍了VCS集群架构、监控与故障切换机制。接着,本文阐述了备份策略的基本原则,备份工具的选择与配置,并提供备份执行与
【Android Studio多屏幕适配指南】:响应式设计的必修课

# 摘要
随着Android设备的多样化,屏幕尺寸和分辨率的差异给开发者带来了多屏幕适配的挑战。本文首先概述了Android Studio多屏幕适配的必要性,并深入探讨了屏幕适配的基础理论,包括屏幕尺寸和分辨率的分类、响应式布局的重要性以及dp与px单位的使用。实践技巧章节提供了使用不同布局资源和高级布局适配技术的具体方法。进阶应用章节讨论了如何处理屏幕方向变化、优化工具使用以及处理不同屏幕密
高级配置指南:基恩士与西门子设备在复杂PROFINET网络中的应用秘籍
# 摘要
随着工业自动化的发展,PROFINET网络因其高效性和可靠性在工业控制系统中得到了广泛应用。本文首先介绍了PROFINET网络的基础知识,然后详细探讨了基恩士设备在该网络中的配置技巧,包括设备功能、参数设置以及安全性维护。接着,文章转向西门子设备的集成方案,阐述了PLC和HMI的配置以及数据交换和监控的重要性。在复杂网络环境下,设备互联的挑战、故障诊断和网络性能优化方法成为研究的重点。最后,本文展望了PROFINET技术的未来发展趋势,包括高级配置技术和与工业物联网的融合。通过对这些主题的深入分析,本文旨在为工程师提供在实践中实施和优化PROFINET网络的实用指南。
# 关键字
【模拟电路故障快速诊断】:专业技巧大公开,快速定位问题
# 摘要
本文旨在探讨模拟电路故障的快速诊断方法,涵盖了从理论基础到实际应用的各个方面。首先介绍了模拟电路的基础理论、常见故障类型及其成因,并着重讲解了故障检测的基本方法。其次,本文提供了实践中的故障诊断技巧、工具选择及案例分析,以及故障预防和维护策略。在此基础上,进一步分析了仿真技术在故障诊断中的应用以及高级诊断技术,包括先进信号分析技术和复
【User Gocator全解析】:2300系列使用手册深度解读(提升技能必备)
# 摘要
本文全面介绍了User Gocator 2300系列的硬件构成、软件操作以及高级应用。第一章概述了User Gocator 2300系列的特点与应用领域。第二章详细分析了该系列的核心硬件组件,包括激光扫描引擎和图像采集系统,以及硬件接口与连接的详细功能,并提供了硬件维护与升级的策略。第三章深入探讨了软件操作方面,从用户界面的布局和功能到软件配置和校准,以及软件调试与优化的最佳实践。第四章则着重于高级应用,涵盖自定义测量工具的创建、数据处理与分析,以及系统集成与自动化测试流程。第五章通过行业应用实例和技术创新解决方案的案例研究,展示了User Gocator 2300系列在不同场景下的
分布式系统性能提升指南:量化因子选择对系统影响的案例研究
# 摘要
本文旨在探讨分布式系统性能影响因素,并着重分析量化因子在性能评估与优化中的关键作用。首先,本文通过理论基础和量化因子的重要性,阐述了量化因子与系统性能之间的关系,并讨论了选择合适量化因子的方法论。随后,本文转向实践应用策略,探讨量化因子的集成、部署,以及如何通过性能监控与量化因子反馈循环进行持续性能优化。在实战章节,详细描述了性能优化流程和量化因子在其中的应用,通过具体案例展示了分布式系统性能提升的实践成效。最后,本文展望了量化因子与未来分布式系统的发展趋势,包括新技术的应用和跨学科研究的深入。整体而言,本文为理解和应用量化因子提供了一个全面的框架,并指出了量化因子在持续改进分布式系
RTL8306E高级编程指南:性能极限挑战与故障解决快速通道
# 摘要
本文系统地介绍了RTL8306E芯片的特性、性能极限挑战、故障诊断与解决方法、高级编程技术以及应用拓展与创新。首先,概述了RTL8306E的基本特性,然后深入探讨了其性能极限的理论基础和测试评估方法,并通过案例分析挑战极限时的实际表现。接着,本文详细阐述了故障诊断的理论与方法,以及常见的故障案例及其解决策略。进一步地,文章揭示了RTL8306E在高级编程技术方面的应用,并提供了有效的开发环境与工具集成解决方案。在应用拓展与创新方面,分析了RTL8306E在不同场景中的性能优化和新兴技术的集成。最后,展望了RTL8306E的未来趋势和其在社区中的潜在贡献。本文旨在为使用RTL8306E
【数据完整性】:Replace与Regexp在数据库维护中的重要性

# 摘要
本文详细探讨了数据完整性维护的关键技术,重点关注Replace语句和Regexp在现代数据库中的应用。首先,本文介绍了Replace语句的基本原理和在数据维护中的高效应用,包括其与Insert和Update语句的对比,以及在批量数据替换和事务处理中的高级技巧。其次,文章深入分析了Rege
【系统迁移与部署】

# 摘要
随着信息技术的快速发展,系统迁移与部署成为企业优化IT架构和提升业务连续性的重要手段。本文详细探讨了系统迁移与部署的理论基础、关键技术、实际操作步骤、部署策略和最佳实践,以及未来趋势。通过对迁移准备、执行过程、风险评估与管理的深入分析,本文章详细阐述了硬件和软件迁移的具体操作,并着重论述了数据同步、系统兼容性分析等关键技术。在部署策略方面
【信号分析与处理精通】:CANoe 10.0精确诊断数据背后的信息

# 摘要
本文深入探讨了CANoe 10.0这一强大的网络分析工具,包括其基础概念、信号分析与处理理论,以及实际应用中的信号分析和处理技术。首先概述了CANoe 10.0的基础知识,并着重分析了信号分析与处理的理论基础,涵盖了信号的定义、分类、分析工具的选择和高级信号处理技术。接着,文中详细阐述了如何在CAN
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )