生物信息学基础:DNA序列数据的获取与处理
发布时间: 2024-02-29 09:36:27 阅读量: 64 订阅数: 23 

生物信息学数据分析 chip-seq
# 1. 导言
## 1.1 介绍生物信息学在现代生物学研究中的重要性
生物信息学在当代生物学研究中起着举足轻重的作用。随着高通量测序技术的发展和广泛应用,生物学领域产生了大量的DNA序列数据,这些数据包含了生物体内DNA的遗传信息。生物信息学通过整合、分析和解释这些数据,为研究者提供了丰富的信息资源,加速了生命科学研究的进展。
## 1.2 引言DNA序列数据在生物信息学中的作用
DNA序列数据是生物信息学的核心内容之一,它包含了生物体内基因的编码信息以及遗传变异的信息。通过对DNA序列数据的分析,研究者们可以揭示基因组的结构与功能、基因间的关联关系、生物的进化历程以及基因与性状之间的关联等重要信息。因此,DNA序列数据在生物信息学中具有重要的地位,对生物学研究具有深远影响。
以上是第一章节的内容,后续章节和代码示例将继续完善。
# 2. DNA序列数据的获取
DNA序列数据的获取是生物信息学研究的第一步,有多种途径可以获取DNA序列数据,包括实验室方法和公共数据库。
### 实验室方法:测序技术简介
在实验室中,科学家通过测序技术可以获得DNA序列数据。随着技术的不断发展,测序技术的速度和准确性得到了显著提升,包括Sanger测序、Next-generation sequencing (NGS)、第三代测序技术等。这些技术的不同之处在于样本处理、数据产出量和成本等方面有所差异。
```python
# 以python代码展示Sanger测序的简单示例
def sanger_sequencing(dna_sample):
sequenced_data = ""
for base in dna_sample:
sequenced_data += sequencer_machine(base)
return sequenced_data
def sequencer_machine(base):
# 模拟测序机器的处理过程
return base
dna_sample = "ATCGGCTA"
sequenced_result = sanger_sequencing(dna_sample)
print(sequenced_result)
```
**总结:** 实验室方法通过各种测序技术获取DNA序列数据。
### 公共数据库:常用的DNA序列数据库介绍
生物信息学领域有许多公共数据库可供科研人员查询和下载DNA序列数据,常用的数据库包括GenBank、EMBL、DDBJ等。这些数据库中包含了大量的基因组数据、转录组数据等,为研究人员提供了丰富的数据资源。
```java
// 以Java代码展示如何在GenBank数据库中下载DNA序列数据
import java.io.BufferedReader;
import java.io.InputStreamReader;
import java.net.URL;
public class GenBankDownloader {
public static void main(String[] args) {
String genbankURL = "https://www.ncbi.nlm.nih.gov/nuccore/NC_000913.3?report=fasta";
try {
URL url = new URL(genbankURL);
BufferedReader reader = new BufferedReader(new InputStreamReader(url.openStream()));
String line;
while ((line = reader.readLine()) != null) {
System.out.println(line);
}
reader.close();
} catch (Exception e) {
e.printStackTrace();
}
}
}
```
**总结:** 公共数据库是获取DNA序列数据的重要来源,科研人员可以从中检索和下载所需的数据。
### 数据下载与存储:获取DNA序列数据的途径与注意事项
下载和存储DNA序列数据时,科研人员需要注意数据的格式、文件大小以及数据的合理管理。同时,数据的备份和安全存储也是非常重要的,以防止数据丢失或损坏。
```go
// 以Go语言示例展示如何下载DNA序列数据并保存到本地文件
package main
import (
"io/ioutil"
"net/http"
"os"
)
func main() {
url := "https://www.ebi.ac.uk/ena/data/view/CP003728&display=fasta"
response, err := http.Get(url)
if err != nil {
panic(err)
}
defer response.Body.Close()
data, err := ioutil.ReadAll(response.Body)
if err != nil {
panic(err)
}
file, err := os.Create("sequence.fasta")
if err != nil {
panic(err)
}
defer file.Close()
file.Write(data)
}
```
**总结:** 下载和存储DNA序列数据时需注意数据的格式、文件大小,同时做好数据备份和安全存储工作。
# 3. DNA序列数据的预处理
DNA序列数据在获取后需要进行一系列预处理步骤,以确保后续分析的准确性和高质量。预处理过程包括数据清理、序列比对和拼接等步骤。
#### 数据清理
数据清理是指对原始DNA序列数据进行去噪和纠错的步骤,以保证后续分析的可靠性。常见的清洗工具包括Trimmomatic、Fastp和Cutadapt等。以下是使用Fastp进行数据清理的Python示例:
```python
import subprocess
# 使用Fastp进行数据清理
def run_fastp(input_file, output_file):
cmd = f"fastp -i {input_file} -o {output_file} -q 20 -u 30"
subprocess.run(cmd, shell=True)
# 调用函数
input_file = "raw_sequence.fastq"
output_file = "clean_sequence.fastq"
run_fastp(input_file, output_file)
```
**代码说明:** 上述代码使用Fastp对原始DNA序列数据进行了质量控制和去除接头过程。其中,参数`-i`指定输入文件,`-o`指定输出文件,`-q`设定质量阈值,`-u`设定最小长度阈值。
#### 序列比对
序列比对是将待分析的DNA序列数据与已知的参考基因组进行比对,以寻找同源性和确定序列的来源。常用的比对工具包括Bowtie2、BWA和BLAST等。以下是使用Bowtie2进行序列比对的示例:
```shell
# 使用Bowtie2进行序列比对
bowtie2-build reference.fasta reference_index
bowtie2 -x reference_index -U clean_sequence.fastq -S aligned_sequence.sam
```
**代码说明:** 上述代码首先利用Bowtie2构建参考基因组的索引,然后对清洗后的DNA序列数据进行比对,生成比对结果文件aligned_sequence.sam。
#### 序列拼接
在基因组学研究中,长DNA序列通常会被分割成若干短片段,需要对这些片段进行拼接。常用的序列拼接工具包括Spades、IDBA-UD和SOAPdenovo等。以下是使用Spades进行序列拼接的示例:
```shell
# 使用Spades进行序列拼接
spades.py -1 forward_reads.fastq -2 reverse_reads.fastq -o assembled_genome
```
**代码说明:** 上述代码利用Spades对前后向读取的DNA序列数据进行拼接,生成组装后的基因组序列文件。
通过上述预处理步骤,可以使原始DNA序列数据经过清洗、比对和拼接处理后达到较高质量、准确性和完整性,为后续的DNA序列数据分析奠定基础。
# 4. DNA序列数据的分析
DNA序列数据的分析是生物信息学中非常重要的一个环节。通过对DNA序列数据进行分析,可以揭示基因的结构与功能,描绘基因的进化历程,对基因进行功能注释等。下面我们将详细介绍DNA序列数据的分析过程以及常用的分析方法。
#### 基本序列分析
在基本序列分析中,常用的方法包括ORF(开放阅读框)预测、碱基组成分析和密码子偏好性分析。
1. **ORF预测**:通过寻找DNA序列中可能的开放阅读框,可以帮助确定基因的位置和可能的编码区域。常见的ORF预测工具包括ORFfinder和Prokka。
2. **碱基组成分析**:对DNA序列中碱基的组成进行统计分析,可以了解碱基的分布情况,对序列的特征有所了解。可以使用BioPython等库进行碱基组成分析。
3. **密码子偏好性分析**:通过分析DNA序列中密码子的使用频率,可以了解不同生物体对特定氨基酸的偏好程度。这对基因的表达调控有重要意义。常用工具包括CodonW和Codon Usage Database。
#### 进化分析
DNA序列的进化分析主要包括系统发育树构建和同源基因家族分析。
1. **系统发育树构建**:通过比较不同物种的DNA序列,可以推断它们之间的进化关系,进而构建系统发育树。常用的系统发育树构建软件包括MEGA、PHYLIP和RAxML。
2. **同源基因家族分析**:通过比对不同物种中的DNA序列,可以找到功能相似的基因并建立它们之间的同源基因家族关系。常用工具包括BLAST和OrthoMCL。
#### 功能注释
DNA序列的功能注释是对基因的功能进行预测和解释,常用的方法包括基因本体论(Gene Ontology)分析和通路富集分析。
1. **基因本体论分析**:通过对基因进行分类、注释,构建基因本体,进而了解基因的功能及其调控网络。常用工具包括GO enrichment analysis和TopGO。
2. **通路富集分析**:通过比对基因组、转录组等数据,分析富集在特定生物学过程中的通路,深入了解基因功能。常用工具包括KEGG和Reactome。
DNA序列数据的分析需要结合生物学背景知识和专业工具进行综合分析,对生物学研究具有重要的意义。
# 5. 生物信息学工具与软件
生物信息学领域涵盖了众多实用工具和软件,用于帮助研究人员在DNA序列数据的处理和分析过程中更高效地完成各种任务。这些工具和软件在生物信息学研究中扮演着至关重要的角色,下面将介绍其中一些常用的生物信息学工具和软件,以及它们的功能和特点。
### 常用的生物信息学工具
1. **BLAST (Basic Local Alignment Search Tool)**
- **简介:** BLAST是用于DNA、RNA或蛋白质序列比对的工具,可用于查找序列间的同源性并估计同源性的程度。
- **使用场景:** 用于快速在数据库中搜索序列相似性,寻找可能的同源基因。
- **代码示例:**
```python
from Bio.Blast import NCBIWWW, NCBIXML
result_handle = NCBIWWW.qblast("blastn", "nt", "ACGTAGCTAGCTAGCATG")
blast_record = NCBIXML.read(result_handle)
```
2. **EMBOSS (The European Molecular Biology Open Software Suite)**
- **简介:** EMBOSS是一个开源的生物信息学软件包,包含了一系列用于序列分析的工具。
- **使用场景:** 用于序列比对、搜索、修剪、转换等序列分析任务。
- **代码示例:**
```shell
embossversion
emboss -seqret protein.fasta -outseq protein.txt
```
3. **NCBI工具**
- **简介:** 美国国家生物技术信息中心(NCBI)提供了许多在线工具和数据库,用于生物信息学研究。
- **使用场景:** 通过NCBI工具可以进行序列比对、数据库搜索、基因功能注释等多项操作。
- **代码示例:**
```python
from Bio import Entrez
Entrez.email = "your_email@example.com"
handle = Entrez.esearch(db="nucleotide", term="BRCA1")
record = Entrez.read(handle)
```
### 生物信息学软件
1. **基因组学软件**
- **示例软件:** Bowtie, BWA
- **功能:** 用于基因组序列比对、SNP检测、重测序等。
2. **转录组学软件**
- **示例软件:** Cufflinks, HISAT2
- **功能:** 用于RNA测序数据的拼接、定量分析、差异表达基因筛选等。
3. **蛋白质组学软件**
- **示例软件:** Mascot, Proteome Discoverer
- **功能:** 用于蛋白质鉴定、结构预测、功能注释等。
以上介绍的工具和软件仅是生物信息学领域中的冰山一角,随着技术的不断进步和生物数据量的增加,预计未来会有更多更强大的生物信息学工具和软件涌现,为研究人员提供更多可能性和便利。
# 6. 应用案例和未来展望
生物信息学在基因功能研究中的应用案例分析
生物信息学在个性化医学、农业技术等领域的前景展望
**生物信息学在基因功能研究中的应用案例分析**
生物信息学在基因功能研究领域发挥着重要作用,为科学家们研究基因的结构、功能及相互关系提供了强大的工具支持。通过分析DNA序列数据,科研人员可以预测基因的编码区域、识别基因间的调控元件以及推断基因与表型之间的关系。例如,利用序列比对工具比对不同物种的基因组,可以揭示基因的保守性和进化过程,为研究基因功能演化提供线索。
另外,基于DNA序列的功能注释工具可以帮助研究人员理解基因的生物学功能,预测编码的蛋白质结构和功能域,从而深入研究基因的生物学意义。这些应用案例不仅有助于揭示基因功能的奥秘,还为疾病的诊断和治疗提供了理论支持。
**生物信息学在个性化医学、农业技术等领域的前景展望**
随着生物信息学技术的不断发展,其在个性化医学、农业技术等领域的应用前景十分广阔。在个性化医学中,通过分析个体的基因组数据,可以实现对疾病的个体化风险评估、药物反应预测及精准医疗的实现。基于生物信息学的癌症基因组学研究,为肿瘤的精准诊断和治疗提供了新的思路。
在农业技术领域,生物信息学技术可应用于作物基因组学、转基因技术以及动植物遗传育种等方面,提高作物的产量和质量,改善动植物资源的遗传性状,为农业可持续发展提供支持。
未来,随着大数据、人工智能等技术的不断融合,生物信息学将在基因功能研究、个性化医学等领域展现更加广阔的应用前景,为人类健康、农业生产等领域带来更多的机遇和挑战。

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
JLINK_V8固件烧录故障全解析:常见问题与快速解决

# 摘要
JLINK_V8作为一种常用的调试工具,其固件烧录过程对于嵌入式系统开发和维护至关重要。本文首先概述了JLINK_V8固件烧录的基础知识,包括工具的功能特点和安装配置流程。随后,文中详细阐述了烧录前的准备、具体步骤和烧录后的验证工作,以及在硬件连接、软件配置及烧录失败中可能遇到的常见问题和解决方案
【Jetson Nano 初识】:掌握边缘计算入门钥匙,开启新世界

# 摘要
本论文介绍了边缘计算的兴起与Jetson Nano这一设备的概况。通过对Jetson Nano的硬件架构进行深入分析,探讨了其核心组件、性能评估以及软硬件支持。同时,本文指导了如何搭建Jetson Nano的开发环境,并集成相关开发库与API。此外,还通过实际案例展示了Jetson Nano在边缘计算中的应用,包括实时图像和音频数
MyBatis-Plus QueryWrapper故障排除手册:解决常见查询问题的快速解决方案
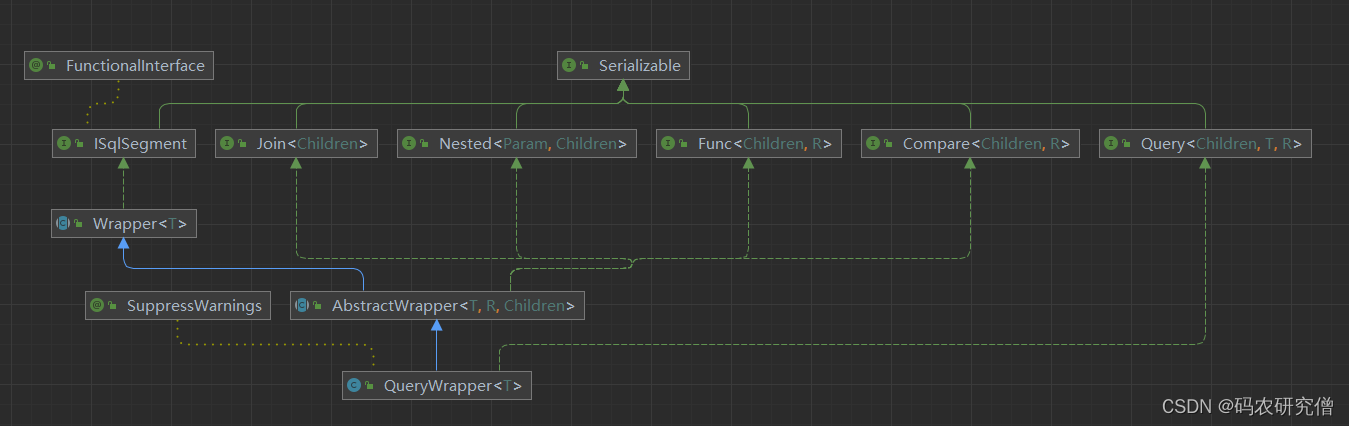
# 摘要
MyBatis-Plus作为一款流行的持久层框架,其提供的QueryWrapper工具极大地简化了数据库查询操作的复杂性。本文首先介绍了MyBatis-Plus和QueryWrapper的基本概念,然后深入解析了QueryWrapper的构建过程、关键方法以及高级特性。接着,文章探讨了在实际应用中查询常见问题的诊断与解决策略,以及在复杂场
【深入分析】SAP BW4HANA数据整合:ETL过程优化策略

# 摘要
SAP BW4HANA作为企业数据仓库的更新迭代版本,提供了改进的数据整合能力,特别是在ETL(抽取、转换、加载)流程方面。本文首先概述了SAP BW4HANA数据整合的基础知识,接着深入探讨了其ETL架构的特点以及集成方法论。在实践技巧方面,本文讨论了数据抽取、转换和加载过程中的优化技术和高级处理方法,以及性能调优策略。文章还着重讲述了ETL过
电子时钟硬件选型精要:嵌入式系统设计要点(硬件配置秘诀)

# 摘要
本文对嵌入式系统与电子时钟的设计和开发进行了综合分析,重点关注核心处理器的选择与评估、时钟显示技术的比较与组件选择、以及输入输出接口与外围设备的集成。首先,概述了嵌入式系统的基本概念和电子时钟的结构特点。接着,对处理器性能指标进行了评估,讨论了功耗管理和扩展性对系统效能和稳定性的重要性。在时钟显示方面,对比了不同显示技术的优劣,并探讨了显示模块设计和电源管理的优化策略。最后,本
【STM8L151电源设计揭秘】:稳定供电的不传之秘

# 摘要
本文对STM8L151微控制器的电源设计进行了全面的探讨,从理论基础到实践应用,再到高级技巧和案例分析,逐步深入。首先概述了STM8L151微控制器的特点和电源需求,随后介绍了电源设计的基础理论,包括电源转换效率和噪声滤波,以及STM8L151的具体电源需求。实践部分详细探讨了适合STM8L151的低压供电解决方案、电源管理策略和外围电源设计。最后,提供了电源设计的高级技巧,包括
NI_Vision视觉软件安装与配置:新手也能一步步轻松入门

# 摘要
本文系统介绍NI_Vision视觉软件的安装、基础操作、高级功能应用、项目案例分析以及未来展望。第一章提供了软件的概述,第二章详细描述了软件的安装流程及其后的配置与验证方法。第三章则深入探讨了NI_Vision的基础操作指南,包括界面布局、图像采集与处理,以及实际应用的演练。第四章着重于高级功能实
【VMware Workstation克隆与快照高效指南】:备份恢复一步到位

# 摘要
VMware Workstation的克隆和快照功能是虚拟化技术中的关键组成部分,对于提高IT环境的备份、恢复和维护效率起着至关重要的作用。本文全面介绍了虚拟机克隆和快照的原理、操作步骤、管理和高级应用,同时探讨了克隆与快照技术在企业备份与恢复中的应用,并对如何
【Cortex R52 TRM文档解读】:探索技术参考手册的奥秘
# 摘要
本文深入探讨了Cortex R52处理器的各个方面,包括其硬件架构、指令集、调试机制、性能分析以及系统集成与优化。文章首先概述了Cortex R52处理器的特点,并解析了其硬件架构的核心设计理念与组件。接着,本文详细解释了处理器的执行模式,内存管理机制,以及指令集的基础和高级特性。在调试与性能分析方面,文章介绍了Cortex R52的调试机制、性能监控技术和测试策略。最后,本文探讨了Cortex R52与外部组件的集成,实时操作系统支持,以及在特定应
西门子G120变频器安装与调试:权威工程师教你如何快速上手

# 摘要
西门子G120变频器在工业自动化领域广泛应用,其性能的稳定性与可靠性对于提高工业生产效率至关重要。本文首先概述了西门子G120变频器的基本原理和主要组件,然后详细介绍了安装前的准备工作,包括环境评估、所需工具和物料的准备。接下来,本文指导了硬件的安装步骤,强调了安装过程中的安全措施,并提供硬件诊断与故障排除的方法。此外,本文阐述了软件配置与调试的流程,包括控制面板操作、参数设置、调试技巧以及性能
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录

文章持续更新中,敬请期待~
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )