揭秘GSEA高级应用:生物信息学数据深度挖掘技巧
发布时间: 2024-12-25 13:27:56 阅读量: 18 订阅数: 17 

数据挖掘 gsea-3.0.jar

# 摘要
基因集富集分析(GSEA)作为一种强大的生物信息学工具,在解读基因表达数据方面发挥着日益重要的作用。本文系统地介绍了GSEA的理论基础、工作机制以及实践操作指南,详细阐述了GSEA在不同生物信息学领域的应用和高级功能拓展,并通过案例实操演示了GSEA的实际应用。文章还讨论了GSEA分析结果的后续处理和验证方法,以及统计学方法在GSEA中的应用。最后,对GSEA未来的发展趋势和挑战进行了展望,强调了机器学习与大数据技术在提升GSEA分析精度和效率方面的潜力。
# 关键字
基因集富集分析;生物信息学;数据分析;案例分析;统计学方法;机器学习
参考资源链接:[GSEA软件使用教程:基因集富集分析详解与数据准备](https://wenku.csdn.net/doc/4pfv1m50q5?spm=1055.2635.3001.10343)
# 1. GSEA在生物信息学中的作用与重要性
生物信息学作为一门飞速发展的学科,其研究过程离不开各种先进的分析方法和技术。基因集富集分析(Gene Set Enrichment Analysis, GSEA)是近年来生物信息学领域内引人注目的分析方法之一。GSEA不仅能够从整体上解释基因表达数据,还能够在统计学意义上识别出与特定生物过程、功能或疾病相关的基因集合。在研究复杂的生物学问题,如癌症、发育生物学等,GSEA提供了更为全面和深入的视角,使得研究者能够更有效地解析实验数据,揭示基因间的潜在联系,进而推动生物信息学向前发展。因此,对于希望深入了解生物信息学分析的应用者来说,掌握GSEA方法不仅是理论需要,也是提高研究能力的重要途径。
# 2. GSEA的基础理论和工作原理
## 2.1 GSEA理论概述
### 2.1.1 GSEA的定义和目的
基因集富集分析(Gene Set Enrichment Analysis,GSEA)是一种用于解析基因表达数据的计算方法,目的是为了评估一个预先定义的基因集合(例如,参与特定生物过程的基因集合)是否在实验数据集中显著富集。该技术与基于单个基因差异表达的分析方法不同,GSEA关注的是基因集合的整体变化趋势。
GSEA通过比较不同实验条件下基因表达的差异来识别与生物过程、通路或功能相关的基因集合的富集情况。这在理解复杂生物数据,尤其是基因表达数据时非常有用,因为它可以揭示在基因整体表达水平上没有显著差异的生物路径的潜在变化。
### 2.1.2 GSEA与传统统计方法的比较
与传统统计方法相比,GSEA不依赖于单个基因的显著性,而是通过统计方法评估整个基因集合的表达模式。在传统方法中,研究人员通常会在实验前后对单个基因进行显著性检验,然后选择具有显著变化的基因进一步分析,这种方法忽略了那些变化幅度不大但协同作用显著的基因集合。
GSEA能够克服一些传统单基因分析的局限性,例如统计功效不足和高假阳性率。它特别适用于基因表达变化不显著的基因集合,或者是基因表达水平变化较为分散的情况,这在许多生物过程中是常见的。此外,GSEA还可以处理高度相关的基因集合,因为它采用了一种称为置换测试的统计方法,通过随机化实验样本的标签来评估基因集合是否富集。
## 2.2 GSEA的工作机制
### 2.2.1 基因集的概念与分类
基因集是由一系列具有相似功能或参与相同生物学过程的基因组成的一个集合。在GSEA分析中,基因集可以是通路、功能、表达模块或任何由一组基因构成的集合。基因集通常来源于公共数据库如KEGG、GO或由研究者根据特定实验目的定义。
GSEA中的基因集根据其来源和特点可以分为不同的类型:
1. **通路基因集**:这些基因集是基于已知的生物通路,如信号通路、代谢通路等。
2. **功能基因集**:这些基因集与特定的生物学功能相关,例如细胞周期、细胞凋亡等。
3. **同源基因集**:基于基因序列相似性或同源关系构建的基因集合。
4. **表达模块基因集**:通过生物信息学分析,如聚类分析,得到的基因集合。
5. **用户自定义基因集**:研究人员基于实验设计或特定研究目标自行定义的基因集合。
### 2.2.2 富集分析的统计模型
GSEA的统计模型基于一个核心假设:功能相关或同通路的基因在生物过程中往往表现出类似的表达模式。因此,GSEA的富集分数(Enrichment Score, ES)反映了一个基因集在整个表达谱中的累积变化。
富集分数的计算基于排序的基因列表,通常按照实验组与对照组之间基因表达差异的统计量(如t检验的p值或差异倍数)进行排序。然后,GSEA计算每个基因集的富集分数,以评估基因集在排序列表中的位置。
计算富集分数的步骤如下:
1. 根据排序列表,计算排名权重。
2. 遍历基因集内的基因,累加排名权重。
3. 将基因集的累加权重与其外的基因进行比较,计算富集分数。
4. 通过置换测试评估富集分数的统计显著性。
### 2.2.3 置换测试和P值计算
置换测试是GSEA的核心步骤,用于评估观察到的基因集富集分数的显著性。置换测试随机改变实验样本的标签,然后重新计算基因集的富集分数,重复这个过程多次(例如1000次),从而得到一个富集分数的经验分布。
基于经验分布,可以计算出观察到的富集分数的p值。p值表示在随机情况下观察到与当前富集分数相当或更极端情况的概率。通常,通过校正多重假设检验(例如使用Benjamini-Hochberg方法),来控制假发现率(False Discovery Rate, FDR)。
p值和FDR的计算对确定基因集是否在实验条件下显著富集至关重要。如果一个基因集的富集分数显著高于随机抽样的情况,那么这个基因集就可以被认为是富集的,表明该基因集中的基因在实验条件的影响下表现出协同作用。
GSEA是一种强大的工具,可以揭示基因表达数据背后的潜在生物学意义。在下一章中,我们将介绍GSEA的实践操作指南,涵盖从软件工具安装到数据分析和结果解读的详细步骤。通过本章节的介绍,读者将获得GSEA理论和方法的全面理解,为之后的实践操作打下坚实的基础。
# 3. GSEA的实践操作指南
## 3.1 GSEA软件与工具安装
### 3.1.1 软件选择与系统要求
在生物信息学研究中,选择合适的软件和工具对于进行基因集富集分析(GSEA)至关重要。GSEA软件由Broad Institute开发,是进行GSEA分析的首选工具。系统要求包括:操作系统需为Windows、Mac OS X或Linux,CPU至少需要一个核心,内存推荐至少有8GB,硬盘空间至少需要数十GB,具体取决于分析数据集的大小。
对于软件选择,GSEA官方提供的是Java编写的GSEA桌面应用,此外还有R语言中的GSEA包以及命令行版本的GSEA,提供给不同用户需求的灵活性。要进行GSEA分析,用户需要下载并安装Java运行环境,然后下载GSEA软件及其必需的数据文件,包括基因集数据库。
### 3.1.2 安装步骤与配置环境
安装GSEA的流程相对简单,以下是详细步骤:
1. 访问Broad Institute的官方网站下载GSEA软件。
2. 根据操作系统选择合适的安装包,并下载。
3. 解压下载的压缩包到指定目录。
4. 运行软件,通常为双击"run"批处理文件(Windows)或脚本文件(Mac/Linux)。
5. 在首次运行时,软件会引导用户下载所需的数据文件。
配置环境主要是指确保运行GSEA所需的JRE(Java Runtime Environment)已经正确安装。用户可以使用GSEA内置的环境检查工具来验证是否所有必需的组件都已正确安装。
## 3.2 GSEA数据准备与预处理
### 3.2.1 数据格式转换和标准化
GSEA的数据输入格式通常是基因表达矩阵,一般包含有基因ID、样本信息以及表达值。常用的数据格式有GCT和RES等,可通过GSEA提供的格式转换工具进行转换。在进行分析之前,数据需要进行标准化处理,以消除不同数据集间的偏差。
例如,可以使用R语言或Python中的相关库(如limma或scikit-learn)来完成数据标准化。标准化的目的是使得样本之间以及基因之间的表达值可以在同一尺度上比较,常用的标准化方法包括z-score标准化、最小-最大标准化等。
### 3.2.2 表达矩阵与基因注释
表达矩阵是GSEA的核心输入文件之一,需要具有准确的基因注释信息。基因注释通常需要包括基因的官方名称、别名以及功能信息,这对于GSEA结果的生物学解释至关重要。GSEA提供了一个名为"chip"的文件格式,用于存储基因集信息和基因注释信息。
确保表达矩阵和基因注释信息正确匹配是至关重要的。在准备数据时,用户需要对数据进行校对,确保每一行的基因ID能够与基因注释文件中的ID相对应。如果有必要,可以使用脚本语言如Python或R进行数据清洗和格式化。
## 3.3 GSEA分析流程详解
### 3.3.1 参数设置与运行分析
GSEA分析流程的设置包括选择合适的基因集数据库、设定适当的统计参数(如置信区间、阈值等),以及调整置换测试的数量和类型。GSEA软件界面提供了丰富的参数选项,允许用户根据研究目的进行个性化配置。
在GSEA软件中,用户需要上传表达矩阵文件、基因集文件以及定义样本类别的标签文件。此外,还可以设置样本抽样方式、排序方式、基因排名方法等。用户需要仔细核对所有参数设置,确保每一步都符合研究要求。
接下来,运行分析。GSEA软件将根据用户设置的参数,执行富集分析,这通常需要一些时间,具体取决于数据集的大小和计算资源。对于大规模数据集或需要进行多次分析的情况,可能需要考虑使用服务器或高性能计算资源。
### 3.3.2 结果解读与可视化
GSEA分析完成后,会生成一系列的输出文件,其中最主要的是一个名为"report"的HTML报告文件。报告文件包含了分析结果的完整解读,包括每个基因集的统计信息、富集图、排名图和关联矩阵等。通过解读这些数据,研究人员可以直观地看到哪些基因集在实验条件间存在显著差异。
为了进一步理解和解释GSEA结果,使用GSEA提供的可视化工具进行结果的图形展示是十分必要的。可视化工具可以帮助研究者识别和展示在数据集中显著富集的基因集。例如,可以利用GSEA软件的内置功能将富集分数、基因排名以及基因集信息整合在一起,生成富集分数曲线图。
此外,还可以使用R语言或Python等编程语言中的相关绘图库(如ggplot2或matplotlib),对GSEA结果进行进一步的自定义绘图和分析。这将有助于研究者深入挖掘数据背后的生物学信息,并将结果与已知的生物学知识进行对比验证。
```mermaid
graph TD;
A[开始GSEA分析] --> B[数据准备]
B --> C[安装GSEA软件]
C --> D[数据格式转换和标准化]
D --> E[表达矩阵与基因注释]
E --> F[设置GSEA参数]
F --> G[运行GSEA分析]
G --> H[解读GSEA结果]
H --> I[结果可视化]
I --> J[保存和分享GSEA报告]
```
在上述流程中,可视化部分尤为关键,因为直观的图形可以极大地促进研究者对数据的感知和理解。GSEA的结果通常包含了基因表达的变化情况、基因集的富集程度以及相应的统计显著性等信息,这些信息被整合在图形中,为生物学假设的提出和进一步实验的设计提供了基础。
# 4. GSEA高级应用与案例分析
## 4.1 GSEA在不同生物信息学领域的应用
### 4.1.1 癌症研究中的应用
在癌症研究中,GSEA(基因集富集分析)被广泛应用于识别与癌症相关的关键生物通路。通过分析肿瘤样本与正常样本之间的基因表达差异,研究人员可以识别出导致癌症进展或抑制的关键基因集。例如,在某项研究中,通过GSEA识别出了与侵袭性表型相关的信号通路,并进一步通过实验验证了这些通路在癌症细胞中的功能性作用。该方法不仅帮助研究人员筛选出与特定癌症表型关联的基因集,也加速了潜在药物靶标的发现过程。
### 4.1.2 干细胞研究中的应用
GSEA在干细胞研究中也发挥着重要作用。通过GSEA分析,研究者们能够理解干细胞分化过程中的基因表达变化和调控机制。例如,研究者通过GSEA分析发现了一组在干细胞分化过程中显著上调的基因集,这些基因集与细胞周期调控密切相关。这一发现有助于理解干细胞如何保持其自我更新和多向分化的能力。此外,GSEA还在识别干细胞治疗相关效果的研究中起到了关键作用,通过分析干细胞治疗前后的表达数据,研究者能够更好地评估治疗效果和潜在的副作用。
## 4.2 GSEA的高级功能拓展
### 4.2.1 多组学数据集成分析
随着生物信息学技术的发展,多组学数据集成分析越来越受到重视。GSEA也逐步融入到这一趋势中,能够处理包括转录组、蛋白质组和代谢组等多组学数据。通过综合分析不同层次的生物数据,GSEA能够提供更为全面的生物学见解。例如,在一项综合转录组和蛋白质组数据的研究中,GSEA不仅揭示了特定基因集在转录水平上的变化,也揭示了蛋白质表达水平上的相似或差异模式。
### 4.2.2 动态GSEA与时间序列分析
动态GSEA是指在时间序列数据上应用GSEA,来跟踪和分析基因表达模式随时间的变化。通过这种方法,研究者可以观察到在特定生物学过程或细胞状态变化中起关键作用的基因集。例如,在细胞分化过程的研究中,动态GSEA帮助揭示了与细胞周期进程相关基因集的表达模式,并预测了细胞命运决定的关键时间点。
## 4.3 GSEA案例实操演示
### 4.3.1 典型案例选取与数据集概述
为了演示GSEA的实际应用,我们选取了一个与糖尿病相关的案例研究。研究者采集了糖尿病患者和健康对照者的基因表达数据,并使用GSEA分析了两组之间的差异。数据集包括数千个基因的表达水平,研究人员首先对数据进行了预处理,包括标准化和归一化处理,以消除实验条件和技术差异带来的影响。
### 4.3.2 案例操作流程与分析结果
在GSEA分析中,研究者关注了与胰岛素信号传导相关的基因集,并发现这些基因集在糖尿病患者中显著下调。分析结果通过GSEA特有的图形和表格形式展示,包括富集分数(ES)、假发现率(FDR)和基因集的排名等信息。这些结果表明,胰岛素信号传导通路可能在糖尿病的发展中扮演重要角色,并为后续的靶向治疗提供了新的研究方向。
```mermaid
graph TD;
A[开始分析] --> B[数据预处理]
B --> C[选择基因集]
C --> D[运行GSEA分析]
D --> E[解读分析结果]
E --> F[结果可视化展示]
F --> G[生物实验验证]
```
以上流程图展示了从数据预处理到结果分析的整个GSEA应用过程。通过这个案例,我们可以看到GSEA作为一个强大的工具,是如何帮助研究者从大数据中提取有意义的生物学信息,并指导后续的实验设计和验证工作。
```markdown
表格形式展示基因集富集分析结果:
| 基因集 | ES | NES | FDR | P-val |
|--------|----|-----|-----|-------|
| Pathway A | 0.65 | 1.92 | 0.015 | 0.032 |
| Pathway B | -0.58 | -1.88 | 0.023 | 0.041 |
| Pathway C | 0.47 | 1.65 | 0.034 | 0.047 |
```
上表列出了部分分析结果,其中ES代表富集分数,NES代表标准化富集分数,FDR代表假发现率,P-val代表P值。这些参数帮助研究者判断基因集富集分析的统计学显著性和生物学意义。通过表格形式的展示,可以直观地比较不同基因集在实验组和对照组中的表达模式变化。
```python
# 示例代码展示GSEA分析的部分步骤
import gseapy as gp
# 加载基因表达数据
data = pd.read_csv('expression_data.csv')
# 运行GSEA分析
gsea_results = gp.prerank(
data=data,
gene_sets='gene_sets.gmt',
permutation_type="phenotype",
outdir="GSEA_results",
min_size=15,
max_size=500,
eset_name="ES",
chip_name="chip_name",
permutation_num=1000,
weighted_score_type=1,
ascending=False,
processes=4
)
# 查看分析结果
print(gsea_results.res2d.head())
```
上述Python代码使用了`gseapy`库来执行GSEA分析。代码首先加载了表达数据,然后定义了基因集文件和分析参数。通过调整参数,可以优化分析过程并获得更准确的结果。最终,代码打印出了分析结果的前几行,为研究者提供了初步的生物学见解。这样的分析可以帮助研究人员识别出在不同生物过程中起关键作用的基因集,从而指导他们进行更深入的研究。
# 5. GSEA分析结果的后续处理与验证
## 5.1 结果验证的必要性与方法
### 5.1.1 结果验证的重要性
GSEA(Gene Set Enrichment Analysis)作为一种强大的生物信息学分析方法,能够帮助研究者从整体上理解大量基因表达数据。通过基因集合的富集分析,GSEA可以揭示特定生物学过程、通路或功能类别在实验条件下的变化趋势。然而,由于生物信息学分析的复杂性和实验数据的局限性,GSEA分析结果需要通过后续的验证步骤来确认其生物学意义和实验的可重复性。
进行GSEA结果验证的必要性主要体现在以下几个方面:
1. **确保结果的可靠性**:由于GSEA涉及到复杂的统计和算法过程,验证可以作为对分析结果的一个质量控制步骤,确保所得结果不是偶然出现的。
2. **强化生物学意义**:将GSEA结果与独立实验(如qPCR、西方印迹等)的结果对比,可以为GSEA揭示的通路或功能类别提供生物学证据。
3. **推动后续研究**:验证结果有助于研究者理解实验数据的生物学背景,指导后续的实验设计和研究方向。
### 5.1.2 采用的生物实验方法
为了验证GSEA分析结果,通常采用以下几种生物实验方法:
1. **定量PCR(qPCR)**:定量PCR是一种广泛使用的技术,用于测量特定基因的mRNA水平,从而对GSEA中的基因表达变化进行验证。
2. **西方印迹(Western Blot)**:这种方法可以用来检测特定蛋白质的表达水平和翻译后修饰,以验证GSEA结果中涉及的信号通路或生物学过程。
3. **免疫组化(IHC)**:此技术用于在组织水平上检测特定蛋白的表达和定位,有助于验证GSEA结果。
4. **免疫荧光(IF)**:免疫荧光可用于观察特定蛋白在细胞中的分布和定位情况,以此对GSEA结果进行进一步的验证。
5. **CRISPR基因编辑**:通过在实验模型中敲除或过表达某个基因,可以验证该基因或其相关通路的功能重要性。
6. **RNA干扰(RNAi)**:使用RNA干扰技术沉默目标基因表达,观察对相关生物学过程的影响,从而对GSEA结果进行功能验证。
## 5.2 统计学方法在GSEA中的应用
### 5.2.1 统计方法的选择
统计学方法在GSEA中的应用,旨在为分析结果提供可靠的证据支持。选择合适的统计方法对于确保研究结论的准确性和可信度至关重要。下面列举了几种常用的统计学方法:
1. **Fisher精确检验**:对于小型基因集或稀有事件的分析,Fisher精确检验是一个有力的工具。
2. **t检验或ANOVA**:当比较两个或多个样本组的均值差异时,t检验或方差分析(ANOVA)可以用来确定差异是否显著。
3. **非参数检验**:如曼-惠特尼U检验或克鲁斯卡尔-瓦利斯检验,适用于数据不满足正态分布假设的情况。
4. **多重测试校正**:当进行多次统计检验时,例如在多个基因集上应用GSEA,需要进行多重测试校正来控制假阳性率。常用的校正方法包括Bonferroni校正和Benjamini-Hochberg程序。
### 5.2.2 假设检验与置信区间
进行假设检验是验证GSEA结果的统计学基础。以下为常见的假设检验与置信区间应用:
1. **原假设与备择假设**:在假设检验中,首先设定原假设(H0),通常表示“无差异”或“无效应”,备择假设(H1)表示研究者希望证明的效应存在。
2. **P值**:P值是在原假设为真的条件下,观察到当前或更极端结果的概率。P值越小,拒绝原假设的证据就越强。
3. **置信区间**:置信区间给出了一个参数的估计范围,这个范围在一定的置信水平(如95%)内包含真实的参数值。如果一个效应的置信区间不包括零或某特定值,那么这个效应可以被认为是统计显著的。
在使用假设检验和置信区间时,研究者应关注以下几点:
- **选择合适的检验方法**:根据数据的分布特性和研究设计选择适当的检验方法。
- **解释P值与效应大小**:P值仅提供统计显著性的证据,而效应大小则说明了效应的实际重要性。两者需要结合分析。
- **注意多重比较问题**:多重比较可能导致假阳性结果的增多,因此需要进行校正。
- **报告详细信息**:在研究论文中,应详细报告统计方法、P值、置信区间和多重比较校正等信息,以便于同行评审和结果的再验证。
# 6. GSEA的未来发展趋势与挑战
## 6.1 GSEA技术的前沿进展
### 6.1.1 机器学习与深度学习在GSEA中的应用
随着人工智能技术的迅速发展,机器学习和深度学习已经开始在GSEA(基因集富集分析)中扮演重要角色。这些先进的分析技术不仅能够处理高维度的数据集,而且能够从数据中学习复杂的模式和关联。
- **机器学习模型:** 如支持向量机(SVM)和随机森林(RF),可以在GSEA中用于特征选择,从而突出与特定生物学过程最相关的基因集。
- **深度学习网络:** 如卷积神经网络(CNN)和循环神经网络(RNN),这些网络能够识别复杂的非线性关系,对基因表达数据进行深层次的特征提取和模式识别。
**示例代码:**
```python
from sklearn.ensemble import RandomForestClassifier
from sklearn.model_selection import train_test_split
from sklearn.metrics import accuracy_score
# 假设 X 是基因表达数据,y 是标签数据
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2)
clf = RandomForestClassifier(n_estimators=100)
clf.fit(X_train, y_train)
predictions = clf.predict(X_test)
print(f"Accuracy: {accuracy_score(y_test, predictions)}")
```
### 6.1.2 大数据分析与云计算平台的结合
在大数据时代,GSEA分析正面临前所未有的数据量挑战。云计算平台如 AWS、Google Cloud 和 Azure 提供了弹性资源和强大的数据处理能力,这对GSEA的发展是极大的推动。
- **弹性计算能力:** 允许按需分配计算资源,确保GSEA分析可以高效地处理海量数据。
- **存储和数据管理:** 提供大规模数据存储方案,并支持高效的数据管理和查询功能。
- **分布式计算框架:** 如Hadoop和Spark,它们支持分布式计算,使GSEA能够并行处理大数据集,显著加快分析速度。
**云服务应用案例:**
```python
# 使用PySpark进行大数据基因集富集分析
from pyspark.sql import SparkSession
spark = SparkSession.builder \
.appName("GSEA on Cloud") \
.getOrCreate()
genes_df = spark.read.csv("path_to_large_genomic_data.csv", header=True)
# 执行基因集富集分析的代码逻辑
spark.stop()
```
## 6.2 GSEA面临的挑战与解决方案
### 6.2.1 数据质量和异质性问题
在GSEA分析中,数据的质量和一致性是非常重要的。高质量的基因表达数据可以提供更可靠的分析结果,但现实情况中数据往往存在异质性。
- **标准化处理:** 对不同来源和平台的数据进行预处理和标准化,以减少数据异质性。
- **数据清洗:** 识别并处理缺失数据、异常值,确保分析的准确性。
**示例处理流程:**
```python
import pandas as pd
# 假设df是未经处理的基因表达数据集
df = pd.read_csv("path_to_raw_genomic_data.csv")
# 进行数据清洗和标准化处理
df_cleaned = df.apply(lambda x: (x-x.mean())/(x.std()) if x.std() != 0 else 0)
df_cleaned = df_cleaned.fillna(0)
# 保存清洗后的数据集
df_cleaned.to_csv("path_to_cleaned_genomic_data.csv", index=False)
```
### 6.2.2 分析标准化和算法优化
GSEA作为一种统计方法,在不同研究中应用广泛,但结果的一致性需要通过标准化的分析流程来保证。
- **标准化流程:** 发展一套通用的GSEA分析流程,减少因分析方法差异导致的结果差异。
- **算法优化:** 优化现有的GSEA算法,以提高统计力并减少假阳性。
**分析流程标准化提案:**
```markdown
1. 数据质量控制和预处理
2. 基因集的选择和准备
3. 参数的选取和优化
4. 分析方法的选择和应用
5. 结果的统计分析和验证
6. 结果的解释和可视化展示
```
通过以上章节的深入分析,我们不仅了解到GSEA作为基因组学研究中的关键工具的当前应用和发展前景,也清晰地看到了面临的主要挑战及其应对策略。这些内容对希望深入生物信息学领域并利用GSEA进行研究的IT专业人员来说,具有较高的参考价值。

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
《GSEA 软件使用教程》专栏是一份全面的指南,涵盖了 GSEA(基因集富集分析)软件的使用。专栏从基础入门到高级应用,提供了一系列深入的教程。读者将学习如何安装和配置 GSEA,准备数据,优化分析参数,进行多重假设校正,并解读分析结果。此外,专栏还介绍了 GSEA 与 R 语言的集成,表型差异分析,KEGG 通路整合,药物研发中的应用,以及高性能计算。通过学习本专栏,读者将掌握 GSEA 软件的全面知识和技能,从而能够有效地进行生物信息学数据分析,探索基因集富集模式,并揭示生物过程的潜在机制。
专栏目录

文章持续更新中,敬请期待~
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
深度解析EDA软件:算法优化让你的设计飞起来

# 摘要
本文全面概述了EDA(电子设计自动化)软件及其在现代电子设计中的核心作用。首先介绍了EDA软件的定义、发展历程和主要分类,然后深入探讨了算法优化的理论背景和实践应用,包括算法复杂度分析、设计策略及优化方法论。接着,文章分析了布局布线、逻辑综合和设计验证优化的实际案例,并讨论了算法优化的高级技巧,如机器学习、多核并行计算和硬件加速技术。通过对EDA软件性能评估指标的分析,本
【管理与监控】:5个关键步骤确保Polycom Trio系统最佳性能

# 摘要
本文全面介绍了Polycom Trio系统的架构、性能评估、配置优化、监控与故障诊断、扩展性实践案例以及持续性能管理。通过对Polycom Trio系统组件和性能指标的深入分析,本文阐述了如何实现系统优化和高效配置。文中详细讨论了监控工具的选择、日志管理策略以及维护检查流程,旨在通过有效的故障诊断和预防性维护来提升系统的稳定性和可靠性。
电力半导体器件选型指南:如何为电力电子项目挑选最佳组件
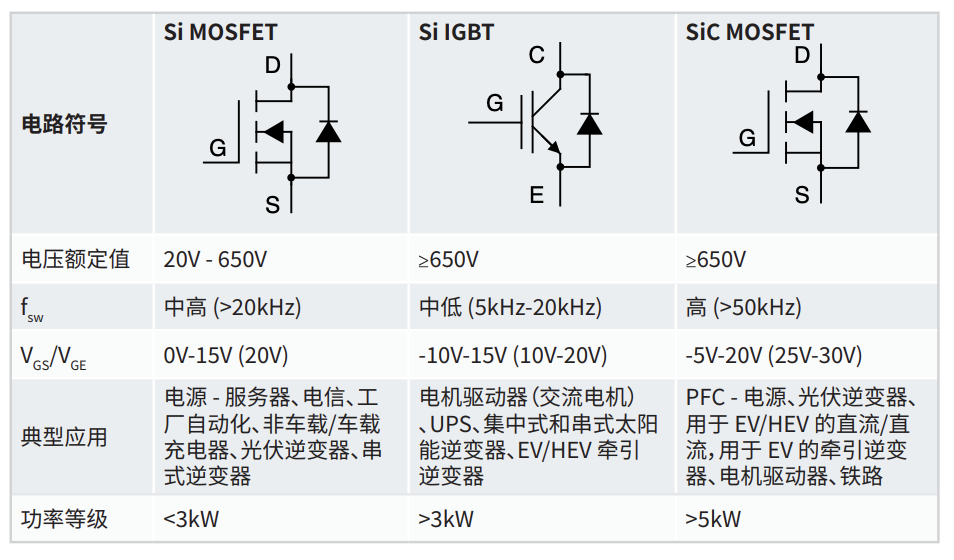
# 摘要
本文全面概述了电力半导体器件的基础知识、技术参数、选型实践考量以及测试与验证流程。在技术参数方面,文章详细介绍了器件的电气特性、热性能和可靠性指标,为电力系统工程师提供了选型时的决策依据。选型实践部分则侧重于应用场景分析、成本效益评估和未来发展考量,旨在指导工程师们在实际工程中做出既经济又可靠的选择。此外,本文还
【mike11建筑模拟全攻略】:从入门到高级应用的全方位教程
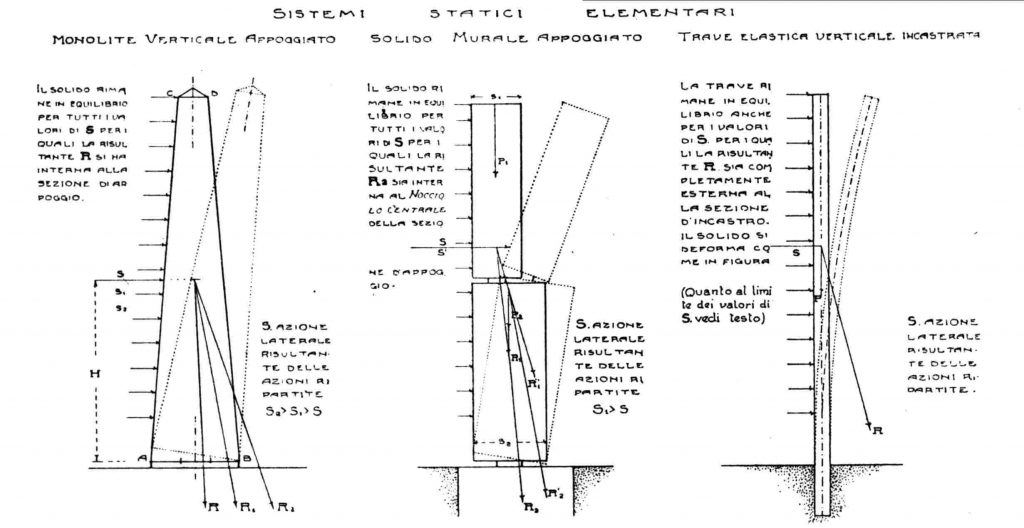
# 摘要
本文全面介绍了mike11建筑模拟软件的各个方面,从基础操作到高级技巧,为建筑模拟提供了一个系统的指导。首先,文章对mike11软件的界面布局、基本设置和视图渲染等基础操作进行了详细介绍。接着,深入探讨了建筑模拟理论基础,包括模拟的目的、建筑物理基础以及模拟流程和参数设置。进阶技巧章节则着重于高级建模技术、环境与气候模拟以及能效与
斯坦福教材揭秘:凸优化理论到实践的快速跨越

# 摘要
本论文系统地介绍了凸优化的基本概念、数学基础、理论框架,以及在工程和科研中的应用案例。首先,文章概述了凸优化的基础知识和数学基础,并详细解析了线性规划、二次规划和对偶理论等关键理论。接着,文章探讨了凸优化工具的使用和环境搭建,强调了模型建立与简化的重要性。随后,通过机器学习、信号处理、运筹学和控制系统等多个领域的应用案例,展示了凸优化技术的实用性。最后,论文展望了凸优化领域的发展趋势,讨论
【tc itch扩展性】:拉伸参数在二次开发中的角色与挑战,稀缺的深入探讨
# 摘要
本文对tcsh shell环境中的参数扩展技术进行了全面的探讨和分析。从参数扩展的基本概念、规则、类别及模式匹配等理论基础出发,深入解析了其在脚本编写、调试优化以及第三方工具集成中的具体应用。文章还着重介绍了复杂参数处理、函数编程中的应用技巧,以及在错误处理中的重要作用。针对二次开发中的挑战,提出了相应的策略和解决方案,并通过案例研究具体分析了参数扩展在特
【网络延迟优化】:揭秘原因并提供实战优化策略

# 摘要
网络延迟是影响数据传输效率和用户体验的关键因素,尤其是在实时性和高要求的网络应用中。本文深入探讨了网络延迟的定义、产生原因、测量方法以及优化策略。从网络结构、设备性能、协议配置到应用层因素,本文详细分析了导致网络延迟的多方面原因。在此基础上,文章提出了一系列实战策略和案例研究,涵盖网络设备升级、协议调整和应用层面的优化,旨在减少延迟和提升网络性能。最后,本文展望了未来技术,如软件定义网络
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
文章持续更新中,敬请期待~
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )