MATLAB求导函数与生物信息学:赋能基因组分析与药物发现,探索生命科学的奥秘
发布时间: 2024-06-14 07:42:15 阅读量: 81 订阅数: 37 

# 1. MATLAB 求导函数概述
MATLAB 求导函数是一种用于计算函数导数的强大工具。导数是函数变化率的度量,在许多科学和工程应用中至关重要。MATLAB 提供了多种求导函数,包括 `gradient`、`diff` 和 `symbolic`。
`gradient` 函数用于计算多变量函数的梯度,即函数在每个维度上的偏导数。`diff` 函数用于计算一维函数的差分,即相邻值之间的差值。`symbolic` 函数用于处理符号表达式,允许以解析方式计算导数。
MATLAB 求导函数的优点包括:
* 易于使用,语法简单明了。
* 计算速度快,即使对于复杂函数也是如此。
* 能够处理各种类型的函数,包括标量、向量和矩阵。
# 2. 求导函数在生物信息学中的应用
求导函数在生物信息学中发挥着至关重要的作用,为各种应用提供了强大的分析和预测能力。
### 2.1 基因组分析中的序列比对和注释
#### 2.1.1 序列比对算法
序列比对是生物信息学中一项基本任务,用于比较两个或多个 DNA 或蛋白质序列之间的相似性。求导函数在序列比对算法中扮演着关键角色,用于计算序列之间的相似性度量。
最常用的序列比对算法之一是 Needleman-Wunsch 算法,它使用动态规划技术来找到两个序列之间的最优比对。该算法通过构建一个相似性矩阵来计算每个序列位置之间的相似性得分,然后使用求导函数来找到矩阵中的最大值,代表最优比对。
```python
def needleman_wunsch(seq1, seq2):
# 初始化相似性矩阵
matrix = [[0 for _ in range(len(seq2) + 1)] for _ in range(len(seq1) + 1)]
# 填充第一行和第一列
for i in range(1, len(seq1) + 1):
matrix[i][0] = -i
for j in range(1, len(seq2) + 1):
matrix[0][j] = -j
# 计算相似性矩阵
for i in range(1, len(seq1) + 1):
for j in range(1, len(seq2) + 1):
if seq1[i - 1] == seq2[j - 1]:
match_score = 1
else:
match_score = -1
matrix[i][j] = max(
matrix[i - 1][j] + -1, # 缺失
matrix[i][j - 1] + -1, # 插入
matrix[i - 1][j - 1] + match_score # 匹配
)
# 查找最大值
max_score = 0
for i in range(len(seq1) + 1):
for j in range(len(seq2) + 1):
if matrix[i][j] > max_score:
max_score = matrix[i][j]
max_i = i
max_j = j
# 回溯找到最优比对
alignment1 = ""
alignment2 = ""
while max_i > 0 and max_j > 0:
if matrix[max_i][max_j] == matrix[max_i - 1][max_j] + -1:
alignment1 += seq1[max_i - 1]
alignment2 += "-"
max_i -= 1
elif matrix[max_i][max_j] == matrix[max_i][max_j - 1] + -1:
alignment1 += "-"
alignment2 += seq2[max_j - 1]
max_j -= 1
else:
alignment1 += seq1[max_i - 1]
alignment2 += seq2[max_j - 1]
max_i -= 1
max_j -= 1
return alignment1[:
```

 百万级
高质量VIP文章无限畅学
百万级
高质量VIP文章无限畅学
 千万级
优质资源任意下载
千万级
优质资源任意下载
 C知道
免费提问 ( 生成式Al产品 )
C知道
免费提问 ( 生成式Al产品 )
0
0
相关推荐

专栏简介
本专栏深入探讨了 MATLAB 求导函数的方方面面,揭示了其背后的数学原理和实现技巧。从基础到进阶,专栏涵盖了求导函数的艺术、实战指南、常见陷阱和误区,以及在科学计算、工程建模、数据分析、图像处理、信号处理、机器学习、深度学习、计算机视觉、自然语言处理、金融建模、生物信息学、气候建模和材料科学等领域的广泛应用。通过比较数值微分和符号微分,专栏帮助读者选择最优解,提升计算效率。此外,专栏还探讨了求导函数在微分方程求解、优化算法和数据分析中的作用,展示了其在解决复杂数学难题和解锁微积分奥秘中的强大功能。
专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )
最新推荐
【电子打印小票的前端实现】:用Electron和Vue实现无缝打印

# 摘要
电子打印小票作为商业交易中不可或缺的一部分,其需求分析和实现对于提升用户体验和商业效率具有重要意义。本文首先介绍了电子打印小票的概念,接着深入探讨了Electron和Vue.js两种前端技术的基础知识及其优势,阐述了如何将这两者结合,以实现高效、响应
【EPLAN Fluid精通秘籍】:基础到高级技巧全覆盖,助你成为行业专家
# 摘要
EPLAN Fluid是针对工程设计的专业软件,旨在提高管道和仪表图(P&ID)的设计效率与质量。本文首先介绍了EPLAN Fluid的基本概念、安装流程以及用户界面的熟悉方法。随后,详细阐述了软件的基本操作,包括绘图工具的使用、项目结构管理以及自动化功能的应用。进一步地,本文通过实例分析,探讨了在复杂项目中如何进行规划实施、设计技巧的运用和数据的高效管理。此外,文章还涉及了高级优化技巧,包括性能调优和高级项目管理策略。最后,本文展望了EPLAN Fluid的未来版本特性及在智能制造中的应用趋势,为工业设计人员提供了全面的技术指南和未来发展方向。
# 关键字
EPLAN Fluid
小红书企业号认证优势大公开:为何认证是品牌成功的关键一步
# 摘要
小红书企业号认证是品牌在小红书平台上的官方标识,代表了企业的权威性和可信度。本文概述了小红书企业号的市场地位和用户画像,分析了企业号与个人账号的区别及其市场意义,并详细解读了认证过程与要求。文章进一步探讨了企业号认证带来的优势,包括提升品牌权威性、拓展功能权限以及商业合作的机会。接着,文章提出了企业号认证后的运营策略,如内容营销、用户互动和数据分析优化。通过对成功认证案例的研究,评估
【用例图与图书馆管理系统的用户交互】:打造直观界面的关键策略
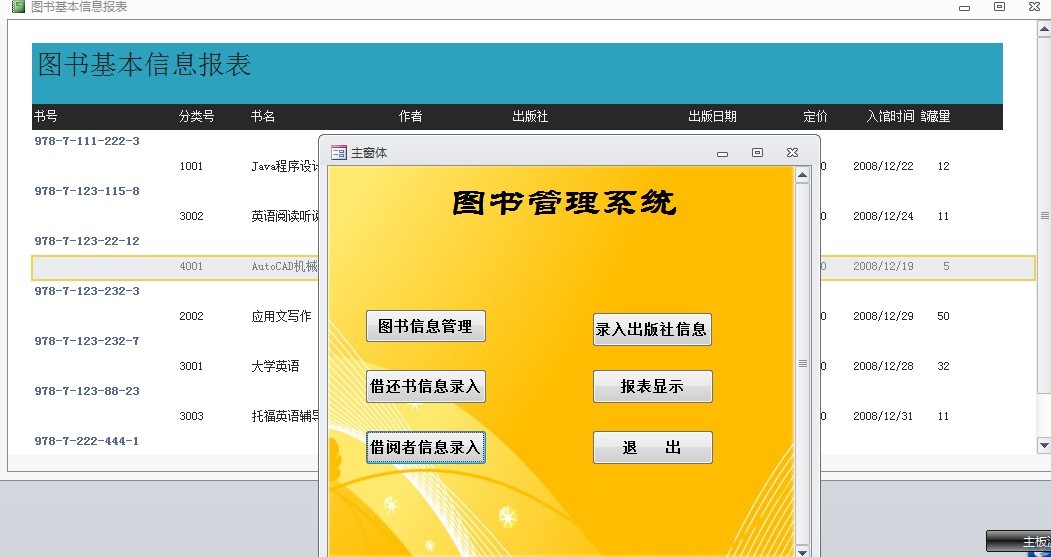
# 摘要
本文旨在探讨用例图在图书馆管理系统设计中的应用,从基础理论到实际应用进行了全面分析。第一章概述了用例图与图书馆管理系统的相关性。第二章详细介绍了用例图的理论基础、绘制方法及优化过程,强调了其在系统分析和设计中的作用。第三章则集中于用户交互设计原则和实现,包括用户界面布局、交互流程设计以及反馈机制。第四章具体阐述了用例图在功能模块划分、用户体验设计以及系统测试中的应用。
FANUC面板按键深度解析:揭秘操作效率提升的关键操作
# 摘要
FANUC面板按键作为工业控制中常见的输入设备,其功能的概述与设计原理对于提高操作效率、确保系统可靠性及用户体验至关重要。本文系统地介绍了FANUC面板按键的设计原理,包括按键布局的人机工程学应用、触觉反馈机制以及电气与机械结构设计。同时,本文也探讨了按键操作技巧、自定义功能设置以及错误处理和维护策略。在应用层面,文章分析了面板按键在教育培训、自动化集成和特殊行业中的优化策略。最后,本文展望了按键未来发展趋势,如人工智能、机器学习、可穿戴技术及远程操作的整合,以及通过案例研究和实战演练来提升实际操作效率和性能调优。
# 关键字
FANUC面板按键;人机工程学;触觉反馈;电气机械结构
华为SUN2000-(33KTL, 40KTL) MODBUS接口安全性分析与防护

# 摘要
本文深入探讨了MODBUS协议在现代工业通信中的基础及应用背景,重点关注SUN2000-(33KTL, 40KTL)设备的MODBUS接口及其安全性。文章首先介绍了MODBUS协议的基础知识和安全性理论,包括安全机制、常见安全威胁、攻击类型、加密技术和认证方法。接着,文章转入实践,分析了部署在SUN2
【高速数据传输】:PRBS的优势与5个应对策略

# 摘要
本文旨在探讨高速数据传输的背景、理论基础、常见问题及其实践策略。首先介绍了高速数据传输的基本概念和背景,然后详细分析了伪随机二进制序列(PRBS)的理论基础及其在数据传输中的优势。文中还探讨了在高速数据传输过程中可能遇到的问题,例如信号衰减、干扰、传输延迟、带宽限制和同步问题,并提供了相应的解决方案。接着,文章提出了一系列实际应用策略,包括PRBS测试、信号处理技术和高效编码技术。最后,通过案例分析,本文展示了PRBS在
【GC4663传感器应用:提升系统性能的秘诀】:案例分析与实战技巧
# 摘要
GC4663传感器是一种先进的检测设备,广泛应用于工业自动化和科研实验领域。本文首先概述了GC4663传感器的基本情况,随后详细介绍了其理论基础,包括工作原理、技术参数、数据采集机制、性能指标如精度、分辨率、响应时间和稳定性。接着,本文分析了GC4663传感器在系统性能优化中的关键作用,包括性能监控、数据处理、系统调优策略。此外,本文还探讨了GC4663传感器在硬件集成、软件接口编程、维护和故障排除方面的
NUMECA并行计算工程应用案例:揭秘性能优化的幕后英雄
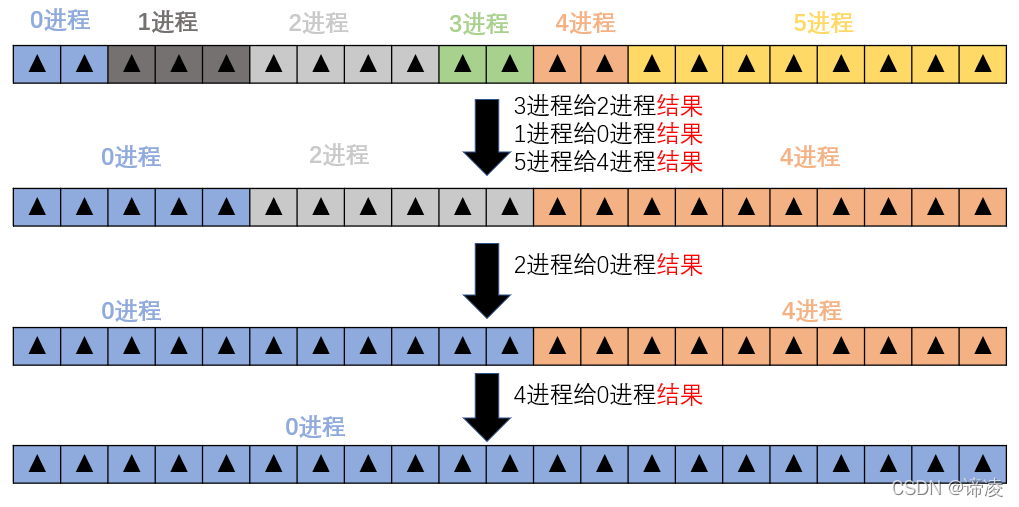
# 摘要
本文全面介绍NUMECA软件在并行计算领域的应用与实践,涵盖并行计算基础理论、软件架构、性能优化理论基础、实践操作、案例工程应用分析,以及并行计算在行业中的应用前景和知识拓展。通过探
资源上传下载、课程学习等过程中有任何疑问或建议,欢迎提出宝贵意见哦~我们会及时处理!
点击此处反馈


专栏目录
最低0.47元/天 解锁专栏
买1年送3月
百万级
高质量VIP文章无限畅学
千万级
优质资源任意下载
C知道
免费提问 ( 生成式Al产品 )